
Abstract
Several editions of the World Health Organization (WHO) classifications of lympho-hemopoietic neoplasms in 2001, 2008 and 2017 served as the international standard for diagnosis. Since the 4th WHO edition, here referred as WHO-HAEM4, significant clinico-pathological, immunophenotypic and molecular advances have been made in the field of lymphomas, contributing to refining diagnostic criteria of several diseases, to upgrade entities previously defined as provisional and to identify new entities. This process has resulted in two recent classifying proposals of lymphoid neoplasms, the International Consensus Classification (ICC) and the 5th edition of the WHO classification (WHO-HAEM5). In this paper, we review and compare the two classifications in terms of diagnostic criteria and entity definition, with focus on mature B-cell neoplasms. The main aim is to provide a tool to facilitate the work of pathologists, hematologists and researchers involved in the diagnosis and treatment of lymphomas.
Similar content being viewed by others

Introduction
Several editions of the World Health Organization (WHO) classifications of lympho-hemopoietic neoplasms in 2001, 2008 and 2016 served as the international standard for diagnosis. However, despite significant progress in the field, there has been no update since the WHO 2016 classification, here referred as WHO-HAEM4. Two classifying proposals of lymphoid neoplasms recently appeared which are hereby named as International Consensus Classification (ICC) and 5th edition of the WHO classification (WHO-HAEM5). The ICC has been published in its definitive format, whilst there only a preview of the WHO Blue Book (which may still be subject to some changes) has appeared. Both ICC and WHO-HAEM5 are based on the same concepts which had inspired the REAL classification [1], e.g., definition of a disease entity based on distinctive pathological, clinical and, when available, molecular features. As compared to WHO-HAEM4 [2], there have been several changes in ICC [3] and WHO-HAEM5 [4] that differ in some structural modifications, terminology and additional/upgrade of new entities defined by clinical, immunophenotypic and molecular data. Unlike ICC, provisional entities are not considered in WHO-HAEM5, whilst some categories regarded as provisional in WHO-HAEM4 have been upgraded to definite entities in both the new classifications. Based on progress in genomic studies, multiple myeloma (MM)/plasma cell myeloma (PCM) has undergone major revisions, to include new distinct cytogenetic entities as reported in ICC. The WHO-HAEM5 also includes a section on “transformations of indolent B-cell lymphoma”, not considered in ICC. In general, diagnostic criteria and recommended ancillary studies have been refined in both classifications. In particular “essential” and “desirable” diagnostic criteria for each entity are discussed in WHO-HAEM5 classification. An important practical issue is how the diagnosis should be reported in everyday work. A reasonable compromise for the time being may be that diagnosis is indicated both according to WHO-HAEM5 and ICC.
For all the above reasons, we here review and compare the two classifications in terms of diagnostic criteria and entity definition, with focus on mature B-cell neoplasms. The main aim is to serve as a tool for pathologists, hematologists and researchers involved in the diagnosis and treatment of lymphomas.
Mature B-cell neoplasms
Chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL)
Diagnostic criteria for CLL/SLL are the same in ICC and WHO-HAEM5 and are based on detection of essential antigens, such as CD19, CD20, CD5, and CD23. Other useful markers include CD43, CD79b, CD81, CD200 and ROR1 [5]. Before starting therapy, the mutational status of IGHV and TP53/17p alterations should also be assessed [6, 7]. Mutations of NOTCH1, SF3B1 and BIRC3 [8] may have a prognostic value [9, 10] but their search remains optional [11]. Complex karyotype and TP53/17p alterations identify high-risk patients [12] who can benefit from targeted therapies [13]. In the WHO-HAEM5, the term “prolymphocytic progression of CLL” has been introduced. Accelerated CLL [14] should be distinguished from diffuse large B-cell transformation (Richter syndrome), that is characterized by sheets of large cells rather than expanded proliferation centers. Richter-like transformation, incidentally observed after ibrutinib interruption [15, 16] may reflect the emergence of a B-cell clone following the sudden release of B-cell receptor signaling inhibition at drug interruption [16]. This proliferation is abrogated by ibrutinib re-introduction [16].
B-cell prolymphocytic leukemia (B-PLL)
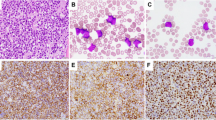
“B-cell prolymphocytic leukemia (B-PLL)” usually occurs in older patients and is characterized by high leukocyte count with >55% prolymphocytes (Fig. 1A), splenomegaly, minimal/absent lymphadenopathy and aggressive course. In the WHO-HAEM5, B-PLL has been deleted as an entity being regarded as a heterogeneous category including cases of hairy cell leukemia variant (HCLv), leukemic mantle cell lymphoma (MCL) and CLL/SLL progressed to B-PLL. Thus, now it has been in part absorbed in the new entity named “splenic B-cell leukemia with prominent nucleoli” (SBLPN) that also includes HCLv (Table 1 and Fig. 2). Conversely, the ICC still regards B-PLL as an entity but recommends its diagnosis only in cases without previous history of B-CLL (to exclude CLL progressing to B-PLL), negative for cyclin D1 and SOX11 (to exclude MCL), and lacking hairy surface projections and intrasinusoidal bone marrow (BM) infiltration (to exclude HCLv and splenic marginal zone lymphoma (SMZL)) (Fig. 1B). B-PLL usually carries a complex karyotype with rearrangement and/or increased copy number of MYC (62%), del17p (38%) and trisomy 18 (30%) [17]. B-PLL patients are treated according to B-CLL guidelines. B-PLL harboring TP53 mutations and/or deletions that predict poor survival usually benefit from bruton tyrosine kinase inhibitors (BTKi) [18, 19]. Patients failing BTKi may still respond to the BCL2 inhibitor venetoclax [20].

A Peripheral blood smear showing typical prolymphocytes (May-Grunwald-Giemsa; ×1000). B Bone marrow trephine showing interstitial infiltration by CD79b positive prolymphocytes (immunoperoxidase staining; ×400). C Imprint from spleen involved by classic mantle cell lymphoma, pleomorphic variant (May-Grunwald-Giemsa; ×400). The tumor cells are medium to large in size and contain evident nucleoli. D Spleen paraffin section from the same case showing a high percentage of Ki-67 positive cells (immunoperoxidase staining; ×400). E Diffuse large B-cell lymphoma of the testis. The asterisk indicates the lumen of a seminiferous tubule (Hematoxylin-eosin; ×400). F The same case as D, showing strong positivity of tumor cells for CD20 (immunoperoxidase staining; 400); the asterisk indicates the lumen of a seminiferous tubule.

B-cell prolymphocytic leukemia (B-PLL), a definite entity in ICC, and hairy cell leukemia variant (HCLv), a provisional entity in ICC, are named in the WHO-HAEM5 under the term of splenic B-cell lymphoma with prominent nucleoli (SBLPN). SBLPN is an heterogeneous category that also comprises cases of unrecognized leukemic mantle cell lymphoma and progressed B-CLL. SDRPL splenic diffuse red pulp small B-cell lymphoma, HCL hairy cell leukemia, SMZL splenic marginal zone lymphoma.
Splenic marginal zone lymphoma (SMZL)
There has been no change in the definition of SMZL in ICC and WHO-HAEM4. SMZL is comprised in the splenic lymphomas/leukemias umbrella of WHO-HAEM5. The diagnosis of SMZL is based on the demonstration of splenomegaly and a BM and/or peripheral blood clonal B-cell population with marginal zone phenotype [21]. The tumor cells carry mutations of KLF2 [22, 23], NOTCH2 [23, 24], TNFAIP3, KMT2D, and TP53, although none of these alterations is specific for SMZL. Search for MYD88 mutations and immunostaining of BM for the myeloid differentiation nuclear antigen [25] may help to distinguish SMZL from lymphoplasmacytic lymphoma (LPL). Sometimes, the splenectomy specimen is mandatory for the histological differential diagnosis.
HCL variant (HCLv)
HCLv is a provisional entity which in WHO-HAEM4 and ICC is grouped within the category of “unclassifiable splenic B-cell lymphomas” together with splenic diffuse red pulp small B-cell lymphoma (SDRPL). HCLv is characterized by marked splenomegaly, lymphocytosis, circulating tumor cells with morphology intermediate between hairy cells and prolymphocytes and lack of monocytopenia. Neoplastic B cells express CD11c and CD103 but not CD25 and annexin A1 [26]. MAP2K1 mutation (p.C121S) has been detected in 7–50% of cases [26, 27] and BRAF V600E is usually absent [26, 28]. In the WHO-HAEM5 [4], HCLv is classified as SBLPN because it is biologically different from classic HCL (HCL) (Table 1 and Fig. 2). In the ICC, HCLv remains a provisional entity. Independently by the terminology (HCLv vs. SBLPN) this disease behaves more aggressively than cHL [29] and is resistant to cladribine alone but sensitive to cladribine or bendamustine plus rituximab. The anti-CD22 immunotoxin [30] is potentially effective [29]. MAP2K1 mutated cases may respond to MEK inhibitors, such as cometinib or trametinib [31].
Splenic diffuse red pulp small B-cell lymphoma (SDRPL)
SDRPL, regarded as an entity both in WHO-HAEM5 and ICC (provisional), is difficult to diagnose because it shares clinical, morphological and immunophenotypic features with other splenic B-cell lymphomas, especially HCLv/SBLPN. Expression of CD200 [32] and cyclin D3 [33] may help in the differential diagnosis. High frequency of BCOR mutations has been detected in SDRPL [34]. However, without splenic tissue demonstrating diffuse infiltration of the red pulp by tumor cells, its distinction from other HCL-like disorders can be very difficult.
Lymphoplasmacytic lymphoma (LPL) and IgM MGUS
Criteria for diagnosis of LPL have been refined. In the WHO-HAEM4, LPL could be diagnosed when clonal lymphoplasmacytic aggregates represented ≥10% of BM cellularity in trephine biopsies. This percentage is confirmed in the WHO-HAEM5 which recognizes two subtypes of LPL: (1) the IgM-LPL/Waldenström Macroglobulinemia (WM) (about 95%); and (2) the non-WM type LPL (about 5%), including cases with IgG or IgA monoclonal proteins, non-secretory LPL, and IgM-LPL without BM involvement. In ICC, LPL can be diagnosed even when clonal lymphoplasmacytic aggregates represent <10% of BM cellularity in trephine biopsies. In general, confident diagnosis of LPL requires recognition of clonal B cells by flow cytometry and clonal plasma cells by immunohistochemistry, and demonstration of abnormal lymphoplasmacytic aggregates by BM trephine biopsy. MYD88 (L265P) is the driver mutation of LPL occurring in about 90% of cases [35] and its search is recommended in both classifications, especially to distinguish LPL from nodal and extranodal MZL [36, 37]. However, the absence of MYD88 mutation does not exclude LPL because a small percentage of cases harbor mutations downstream of MYD88 in the NF-kB signaling pathway [38, 39]. CXCR4 mutations [40] are found in about 40% of cases and associate with symptomatic hyperviscosity and resistance to ibrutinib [41].
In the WHO-HAEM4, HAEM5 and ICC the diagnosis of IgM monoclonal gammopathy of undetermined significance (MGUS) is made in cases showing <10% of BM neoplastic cells with lymphoplasmacytoid or plasma cell differentiation without lymphoplasmacytic B-cell aggregates diagnostic of LPL [42]. Unlike WHO-HAEM5, the ICC recognizes two IgM MGUS entities [3]: (1) the IgM MGUS of plasma cell type; and (2) the IgM MGUS, not otherwise specified (NOS) (Table 1). The first, represents a precursor of IgM MM and it is characterized by the proliferation of clonal plasma cells without B cells and by the absence of MYD88 mutation. The t(11;14) IGH::CCND1 or other myeloma-associated IGH rearrangements may be present. Conversely, IgM MGUS, NOS is characterized by a proliferation of monoclonal B cells (without lymphoplasmacytic aggregates diagnostic of LPL) usually harboring the MYD88 mutation. IgM MGUS, NOS may transform into LPL.
Both the ICC and WHO-HAEM5 now recognize primary cold agglutinin disease (CAD) as an entity distinct from LPL or IgM MGUS (Table 1). CAD is a very rare disorder, more frequent in colder countries [43], which is characterized by the proliferation of clonal B cells producing monoclonal cold agglutinins that mediate an autoimmune hemolytic anemia. CAD typically lacks the MYD88 mutation but displays trisomies of chromosomes 3, 12 and 18 [44], as well as KMT2D and CARD11 mutations [45].
Extranodal marginal zone lymphoma (MZL) of MALT and nodal MZL
Diagnostic criteria for extranodal and nodal MZL are unchanged. Genetic characteristics of extranodal MZL greatly differ depending on the anatomical site [46]. Therapy also varies among anatomical sites, e.g., gastric MALT may benefit from anti-microbial treatment. Despite these differences, neither ICC nor WHO-HAEM5 classify extranodal MZL based on the site of presentation, the only exception being cutaneous MZL (see below). Nodal MZL is a clinically and molecularly heterogeneous entity. Immunostaining for IRTA1 [47] may help in the diagnosis [48, 49]. Recognition of large cell transformation of MZL is based on the detection of aggregates of large B cells.
Pediatric nodal marginal zone lymphoma (PNMZL) considered in the WHO-HAEM4 and in ICC [3] as provisional entity is upgraded in WHO-HAEM5 [4] to a distinct entity. It usually presents with localized disease and can be rarely observed even in adults. Surgical excision is curative in most patients [50]. Distinction of PNMZL from reactive conditions may require demonstration of clonal IG gene rearrangements. Cases with overlapping features between PNMZL and pediatric-type follicular lymphoma have been reported [50].
Both the WHO-HAEM5 [4] and ICC [3] recognize “primary cutaneous marginal zone lymphoma (PCMZL)” as a distinct entity (Table 1). PCMZL resembles morphologically and immunophenotypically nodal and extranodal MZL. However, gain of 6p and loss of 6q (as observed in MZL of ocular adnexa) or the presence of BIRC3-MALT1 fusions (as seen in gastric and pulmonary MZL) are not found. In ICC, PCMZL is called “primary cutaneous marginal zone lymphoproliferative disorder” [3] rather than lymphoma because of its slow growing characteristics, low propensity for systemic dissemination, and good survival with conservative therapy, although cutaneous recurrences are frequent. The WHO-HAEM5 continues to use the term “PCMZL” [4] and to group it among MZL.
Two subtypes of PCMZL/lymphoproliferative disorder are recognized [51]. About 75% of cases express IgG, especially IgG4 (about 50% of cases) [52, 53] without association with pre-existing systemic IgG4-related disease [52]. Conversely, non-cutaneous MZL only rarely express IgG4. This subtype is characterized by the presence of abundant reactive helper type-2 T cells [54], plasma cells located at the periphery of the lymphoid infiltrate and B cells lacking CXCR3 expression. About 25% of cases show a MALT-lymphoma-like IgM+ phenotype, a T-helper type 1-driven process that occurs in older individuals, involves the subcutis, causes follicular colonization and lacks MYD88 mutation. In this setting, non-cutaneous primary disease should be excluded. The molecule IRTA1 [47, 49] may help in identifying these cases [55]. About 63.2% of PCMZL/lymphoproliferative disorder carry dominant-negative mutations involving the death domain of the apoptosis-regulating FAS/CD95 protein [56]. FAS mutations point to an apoptosis defect that may explain the slow growth and indolent course of this tumor [56]. Mutations of SLAMF1, SPEN, and NCOR2 genes have also been reported [56].
Follicular lymphoma (FL)
The WHO-HAEM5 describes three morphological variants of FL: (1) classic FL characterized by follicular growth of centrocytes and centroblasts and t(14;18)(q32;q21)/IGH::BCL2 fusion; (2) follicular large B-cell lymphoma corresponding to the WHO-HAEM4 FL grade 3B that rarely carries the BCL2 translocation. The follicular pattern often co-exists with area of DLBCL. When MUM1/IRF4 is strongly expressed, FISH analysis for IRF4 is required to exclude large B-cell lymphoma with IRF4 rearrangement; and (3) FL with uncommon features comprising FL with “blastoid” or “large centrocyte” features characterized by high proliferative index, and MUM1/IRF4 expression [57] that needs to be distinguished from large B-cell lymphoma with IRF4 rearrangement [58]. FL with predominantly diffuse growth pattern [59] is considered a subtype of classic FL that frequently occurs in the inguinal region and likely corresponds to the ICC entity named BCL2-R-negative, CD23-positive follicle center lymphoma [3] (see below).
In the ICC, the consensus was to retain the morphological grading of FL, as defined in the WHO-HAEM4, although it remains unclear whether clinically the grade 3A differs from grades 1 and 2 [60]. Conversely, patients with grade 3B FL are usually treated as DLBCL [61, 62]. Hence, distinction between grade 3A and 3B is critical [57]. BCL2-rearrangement and CD10 positivity both favor FL grade 3A. In WHO-HAEM5, grading of FL, is regarded as optional because of its scarse reproducibility [63] and questionable clinical significance [64]. The WHO-HAEM4 entity named in situ follicular neoplasia remain unchanged in ICC classification, while in the WHO-HAEM5 it is named in situ follicular B-cell neoplasm. Primary cutaneous follicular lymphoma and duodenal follicular lymphoma remain unchanged in the WHO-HAEM5 and ICC classifications. Pediatric-type FL also remains a distinct entity that usually presents as localized disease in the neck lymph nodes, mostly in young males, being characterized by blastoid cytology, high proliferation rate, lack of BCL2 protein and t(14;18) [65]. MAPK pathway mutations are frequent in pediatric-type FL and the prognosis is excellent following conservative management [65,66,67]. Distinguishing this entity from FL grade 3B remains critical.
Compared to WHO-HAEM4, there are two changes in the ICC but not in the WHO-HAEM5 concerning FL: (1) testicular FL is recognized as a distinct form of FL in young boys; and (2) BCL2-R-negative, CD23-positive follicle center lymphoma [3] is recognized as provisional entity (Table 1). Testicular FL shares clinico-pathological features with pediatric-type FL [68, 69]. In particular, it occurs in children, is limited to testis and usually shows a grade 3 [69]. It also lacks BCL2 expression at immunohistochemistry and BCL2 rearrangements at FISH. Resection, sometimes followed by two cycles of chemotherapy, results in an event-free survival of 100% at a median follow-up of 73 months [69].
Follicle center lymphoma lacking BCL2 rearrangement and expressing CD23 (a surrogate for STAT6 mutations) [3] can be diffuse and purely follicular (about one-third of cases). Although it often presents in the inguinal region, it may also occur in axillary and cervical regions as limited stage disease with favorable prognosis. As opposed to conventional FL, it often presents in women. Molecularly, it shows absence of IGH::BCL2 fusion, and carries frequent STAT6 and CREBBP mutations [59] along with 1p36 deletion [70] or TNFRSF14 mutations [71].
Routine molecular testing is not necessary in FL but advisable under certain circumstances. MAP2K1 mutations favor a diagnosis of pediatric-type FL [67]. Detection of EZH2 mutations is important when therapy with an EZH2 inhibitor is considered [72]. Integration of mutational analysis (m7-FLIPI) in the risk stratification of FL remains investigational [73]. The activity of CAR-T cells [74] against FL is also independent by the underlying genetic landscape.
Mantle cell lymphoma (MCL)
Both ICC and WHO-HAEM5 subdivide MCL into: (1) in situ MCL neoplasm (WHO-HAEM5)/in situ mantle cell neoplasia (ICC); (2) conventional (cMCL); and (3) leukemic non-nodal MCL (Table 1). In situ mantle cell neoplasm/neoplasia represents an incidental finding characterized by colonization of mantle zone of B-cell follicles by neoplastic B cells carrying the IGH::CCND1 fusion and overexpressing cyclin D1 at immunohistochemistry.
cMCL is the most common variant. The IGH::CCND1 fusion generated by t(11;14)(q13;q32) is the genetic hallmark of MCL that is detectable in >95% of cases, IGK or IGL serving as fusion partners in rare cases. Definition of cMCL has been expanded to include cases lacking CCND1 rearrangements but harboring cryptic rearrangements of IGK or IGL enhancers with CCND1 or translocations involving CCND2 or CCND3 [75,76,77,78]. The latter are better detected by FISH or mRNA overexpression than immunohistochemistry. CCND1 rearrangement can also be found in DLBCL raising problems in the differential diagnosis with MCL of plemorphic type. These cases should not be classified as cMCL. Conversely, MYC may be rearranged in bona fide cMCL, usually with blastoid/pleomorphic morphology [79].
Clinically staging of MCL is based on simplified or combination Mantle Cell Lymphoma International Prognostic Index [80, 81]. In the era of new targeted therapies, including BTKi [82, 83] and CD19-directed CAR-T cells [81, 84], every case of MCL should be investigated prior therapy for morphology (blastoid/pleomorphic variant) (Fig. 1C), immunophenotype, proliferative index (labeling of Ki67) (Fig. 1D) and presence of TP53 deletions/mutations since the occurrence of one or more of these variables associates with a poor prognosis [81, 85]. Notably, TP53 deleted/mutated cases are usually resistant to chemotherapy but are sensitive to BTKi [82, 83] or CAR-T cells [81]. Over time, neoplastic cells may acquire mutations conferring resistance to BTKi or venetoclax [86]. Searching these mutations is useful to further guide therapy. Genomic studies point to a high complexity of MCL [87] with possible prognostic impact but additional studies are required before this information is incorporated into classifications.
Leukemic non-nodal MCL is an indolent disease characterized by involvement of BM, spleen and peripheral blood by low proliferating tumor cells and an outcome more favorable than that of cMCL [81]. The diagnosis is favored by the finding of no/limited stage nodal disease, no/low SOX11 expression [88,89,90], high load of somatic IGHV hypermutations (>98%) [89, 91] and lack of ATM mutations/deletions or TP53 mutations [87, 88]. Non-nodal MCL can progress to aggressive cMCL with rapidly enlarging lymphadenopathy following acquisition of TP53 and/or ATM mutations/deletions that confer a poor outcome.
Transformations of indolent B-cell lymphomas
This entity is only considered in WHO-HAEM5 but not in ICC. The term transformation refers to the emergence, following acquisition of additional genetic aberrations, of an aggressive lymphoma in a patient with previously or synchronously diagnosed, clonally-related low-grade B-cell lymphoma [4] (e.g., Richter transformation, transition from FL or MZL to DLBCL). Usually the transformed lymphoma cells retain the immunophenotype of their low-grade counterparts. Transformed lymphomas can manifest as either nodal and/or extranodal disease. The suspicion of transformation can be heralded by enlarging lymph nodes, increasing LDH and/or appearance of systemic symptoms. Observation of the tissue architecture is critical to establish the diagnosis of transformation. Thus, the fine needle aspiration biopsy is not adequate for this purpose.
Diffuse large B-cell lymphoma (DLBCL)
DLBCL not otherwise specified (NOS) represents a heterogeneous entity in terms of morphological features (e.g., centroblastic, immunoblastic), immunophenotype (e.g., CD5+, double MYC/BCL2 expressor) [92,93,94] and cytogenetic/molecular categories [95]. When the diffuse growth pattern cannot be recognized (e.g., fluid-overload-associated large B-cell lymphoma), the WHO-HAEM5 prefers the term of “large B-cell lymphoma”. Most DLBCL, NOS recapitulate the differentiation/maturation processes occurring within the germinal centers (GCs). Cases whose cell-of-origin (COO) is from GC (GCB subtype) show unique gene expression profile [96] and express GC markers at immunohistochemistry. They are also enriched for IGH::BCL2 fusion and mutations of genes involved in GC development, such as EZH2, GNA13, MEF2B, KMT2D, TNFRSF14, B2M and CREBBP [95]. DLBCL deriving from B cells that exit from the GC or from post-GC B cells (activated B-cell-like [ABC] subtype), are characterized by dependence on BCR and NFκB signaling, IRF4/MUM1 expression [97] and enrichment for BCR pathway mutations (e.g., MYD88, CD79B, PIM1) and PRDM1/BLIMP1 mutations/deletions [95].
Both ICC and WHO-HAEM5 recommend the COO of DLBCL should be retained due to its potential prognostic impact [98]. Because of its simplicity, rapidity and low cost, immunohistochemistry is currently the most widely used method in routine practice. However, it cannot recognize the “unclassified” GEP category. In particular, the application of immunohistochemical algorithms [98] to upfront clinical trials of DLBCL, NOS incorporating targeted agents such as bortezomib and ibrutinib have led to disappointing results [99, 100], with the exception of lenalidomide [101]. Thus, COO does not appear to fully capture the high biological complexity of DLBCL. This underlines the importance of moving to a more molecularly-based subdivision of DLBCL. This approach recently led to identifying new genetic subgroups of DLBCL [102,103,104]. However, the functional significance of the driver mutations defining these clusters and their impact on outcome and targeted therapy remains uncertain. Hopefully, in the future such a molecular information may be combined with COO to improve the risk stratification of DLBCL patients for clinical trials [105]. The activity of anti-CD19 CAR-T cells in relapsed/refractory DLBCL [106] seems to be independent from COO and molecular category.
Primary DLBCL of the central nervous system (PCNSL) and primary DLBCL of testis share many features, including the ABC COO and the high frequency of MYD88 and CD79b mutations [105, 107,108,109]. Similarly to CNS, the testis is an immune-privileged site where tumor-infiltrating host immune cells play an important role in immune evasion [110, 111]. Moreover, mechanisms defending the developing gametocytes in the immune-privileged site of the testis may provide DLBCL with an ideal milieu for acquiring an immune-escape phenotype [112]. Thus, primary DLBCL of the testis is now considered by ICC as a distinct entity (Fig. 1E, F). Moving from the same concept, WHO-HAEM5 adopted the term of Large B-cell lymphomas of immune-privileged sites to group cases arising in immune sanctuaries defined by the blood-brain, blood-retinal and blood-testicular barriers [107] (Table 1). Interestingly, CD19-directed CAR-T cells were shown to cross the blood-brain barrier [113, 114]. Primary testicular DLBCL is a rare entity characterized by an aggressive course and tendency to relapse in the controlateral testis and CNS [115]. Primary DLBCL, leg and breast type, intravascular large B-cell lymphoma, and primary adrenal lymphomas show features similar to lymphomas of CNS and testis [116,117,118]. However, ICC and WHO-HAEM5 felt that lumping these tumors under the umbrella term of “extranodal lymphoma ABC type” was premature since they are rather heterogeneous.
The WHO-HAEM4 provisional entity “Burkitt-like lymphoma with 11q aberration” was named as such because of its clinical, morphological (“starry sky pattern”) and immunophenotypic (CD10+/BCL6+/BCL2−/Ki67 high) resemblance to Burkitt lymphoma but lack of MYC rearrangement [119,120,121]. These cases are characterized by 11q aberration, often consisting of a gain in 11q23.2–23.3 followed by a telomeric loss in 11q24.1-qter. Subsequently, they were found to carry DLBCL-associated mutations (e.g., GNA13) without the hallmark Burkitt lymphoma mutations involving ID3-TCF3 or the SWI/SNF complexes [122, 123]. Due to molecular studies showing that this entity is closer to DLBCL than to Burkitt lymphoma, the ICC recognizes it as provisional entity, under the term of “large B-cell lymphoma with 11q aberration” (Fig. 3). Conversely, because of the frequent intermediate/blastoid morphology with starry sky macrophages [124], the WHO-HAEM5 classifies it as “high-grade B-cell lymphoma with 11q aberration” [125]. Chromosome 11q gains/losses can be detected using FISH but chromosomal microarray should be performed if FISH is equivocal. The best therapeutic option for these cases remains uncertain. In pediatric patients, large/high-grade B-cell lymphoma with 11q aberration is less frequent than MYC breakpoint-positive Burkitt lymphoma, occurs at older age, shows less male predominance, lower LDH, less abdominal involvement and it is characterized by an excellent prognosis.

A Lymph node imprint showing large-size tumor cells with basophilic cytoplasm and round nuclei with evident nucleoli (May-Grunwald-Giemsa; ×400). B Tumor cells are double stained for CD20 (green) and BCL6 (brown). C, D FISH reveals 11q aberrations.
Large B-cell lymphoma with IRF4 rearrangement was introduced in the WHO-HAEM4 as a provisional entity under follicular lymphoma and is retained there in the ICC. In the WHO-HAEM5 has been moved, most probably because of the name, to the group of aggressive lymphomas, although this presents in children and young adults with localized disease and excellent outcome [126]. The diagnosis is based on demonstration of IRF4 rearrangement at FISH analysis. IRF4 mutations can also be detected [126]. This entity does not include aggressive B-cell lymphomas with IRF4 rearrangements that also harbor BCL2 and/or MYC rearrangements.
Large B-cell lymphoproliferative disorders related to viral agents according to ICC
The EBV-positive polymorphic B-cell lymphoproliferative disorder, NOS has been added as new entity in ICC. This term should be reserved to cases with/without immunodeficiency showing an alteration of lymph node architecture due to an EBV-positive polymorphic infiltrate that does not fulfill the criteria for diagnosis of lymphoma [127, 128]. In samples without distortion of the nodal architecture by EBV-positive B cells, the term EBV reactivation is recommended [3]. This entity differs from EBV-positive DLBCL, NOS defined by >80% EBV+ cells that can occur at any age and shows a wide morphological spectrum ranging from monomorphic to polymorphic and an aggressive clinical course [129, 130]. Separating this entity from EBV+ classic Hodgkin lymphoma (cHL) may be problematic. Demonstration of >1 B-cell marker in a significant percentage of tumor cells, extranodal presentation and/or EBV latency III favors the diagnosis of EBV-positive DLBCL, NOS.
EBV-positive mucocutaneous ulcer (EBVMCU) [131,132,133,134] has been upgraded from provisional to distinct entity in ICC [131]. The WHO-HAEM5 includes this entity and the EBV-positive polymorphic B-cell lymphoproliferative disorder, NOS in the “Lymphoid proliferations and lymphoma associated with immune deficiency and dysregulation” (Table 1). EBCMCU usually presents in elderly (median age 71 years) as unifocal cutaneous or mucosal ulcer (frequently in the oropharyngeal mucosa probably due to the release of virus into saliva) without associated lymphadenopathy or systemic symptoms [134]. Involvement of the gastrointestinal tract is less frequent and may be preceded by an inflammatory bowel disease [135]. EBVMCU often occurs after a local trauma including tooth extraction and develops in immunocompromised patients, e.g., after therapy with methotrexate [133, 136] or in organ transplant recipients [137]. EBVMCU has a wide morphological spectrum and may simulate DLBCL or even cHL. Focal necrosis and angioinvasion are frequently seen. When >2 skin lesions are present, the term EBV-positive B-cell polymorphic LPD, or if appropriate, EBV-positive DLBCL, NOS should be applied [133]. EBV DNA is usually not detectable in the serum despite the biopsies showing strong EBER-ISH positivity of atypical B cells [137]. EBVMCU is usually characterized by spontaneous regressions or remission after discontinuation or dose reduction of immunosuppressive therapy [132]. However, rituximab and rarely chemotherapy may be required to induce remission [134]. Unlike EBV-positive DLBCL, NOS the prognosis is very good.
Immunodeficiency-associated lymphoproliferative disorders according to WHO-HAEM5
The WHO-HAEM5 introduces the term immunodeficiency and dysregulation associated lymphoproliferative disorders (IDD-LPSDs) that considers the following aspects for determining an integrated nomenclature [4]: (1) histological diagnosis (hyperplasia, polymorphic LPD, lymphoma as for immunocompetent patients); (2) presence or not of virus (EBV, KSHV/HHV8); (3) clinical setting/immunodeficiency background (post-transplant, HIV, iatrogenic/autoimmune); and (4) inborn errors of immunity. EBV-positive polymorphic B-cell lymphoproliferative disorder, NOS according to ICC and the EBV+ mucocutaneous ulcer are grouped herein. The relationships with other entities of WHO-HAEM 4 and ICC are shown in Table 1.
The new entity defined as “HHV-8- and EBV-negative primary effusion-based lymphoma” provisional in ICC and “Fluid overload-associated large B-cell lymphoma” defined in WHO-HAEM5 occurs in older males without underlying immunodeficiency and presents with exclusive involvement of body cavities, usually pleura [138, 139]. Patients usually have an underlying pathological condition leading to fluid overload, such as chronic heart failure, renal failure, protein-losing enteropathy or liver failure/cirrhosis. Fluid overload is thought to lead to lymphoma through chronic serosal stimulation. Most cases have been reported from Japan and frequently had a history of hepatitis C infection [138]. It often shows a centroblastic rather than plasmablastic morphology, with expression of at least one B-cell antigen and a GCB COO. It exhibits better prognosis than primary effusion lymphoma, with spontaneous regression or cure following drainage alone [140]. Unlike WHO-HAEM5, the ICC does not accept EBV+ lesions since they are usually associated with immunosuppression and therefore belong to a different category. The fibrin-associated large B-cell lymphoma, regarded in the WHO-HAEM4 and ICC as a subtype of DLBCL-associated with chronic inflammation, is now upgraded to a definite entity in the WHO-HAEM5.
High-grade B-cell lymphomas (HGBCL)
The WHO-HAEM4 included: (1) HGBCL with MYC and BCL2 and/or BCL6 rearrangements (“double-hit” or “triple-hit”); and (2) HGBCL, NOS. In ICC, “double-hit” HGBCL now comprises two entities: (1) HGBCL with MYC and BCL2 rearrangements (with or without BCL6 rearrangement) (HGBCL-DH-BCL2); and (2) a provisional entity named “HGBCL with MYC and BCL6 rearrangements (HGBCL-DH-BCL6)” (Table 1). In WHO-HAEM5, tumors with MYC and BCL2 rearrangements are named diffuse large B-cell lymphoma/high-grade B-cell lymphoma with MYC and BCL2 rearrangements (DLBCL/HGBCL MYC/BCL2).
DLBCL or HGBCL with MYC and BCL2 rearrangements show a morphology ranging from large cells to blastoid/intermediate. FISH break apart probes are recommended for diagnosis but they may miss the rearrangement in up to 20% of cases. This entity is sometimes preceded by a history of FL and shows a gene expression profile close to centroblasts of the GC dark zone [104, 141, 142]. Moreover, it exhibits a mutational signature more similar to FL (CREBBP, BCL2, KMT2D, MYC, EZH2 and FOXO1 mutations) than to DLBCL, NOS (GCB subtype) [143]. Thus, it differs biologically from HGBCL-DH-BCL6 [144]. It also frequently carries MYC hotspot mutations affecting the phosphorylation site and its adjacent amino acids, which are important for MYC protein degradation, resulting in higher MYC expression that may be responsible for the aggressive clinical behavior, [142, 145]. Cases carrying MYC and BCL2 abnormalities other than typical translocations [146] should not be currently classified as DLBCL- or HGBCL-DH-BCL2.
HGBCL-DH-BCL6 is characterized by frequent involvement of extranodal sites, aggressive clinical course and poor prognosis [144, 147]. As compared to DLBCL or HGBCL with MYC and BCL2 rearrangements, it shows GCB immunophenotype less often, is more likely to be CD10(−)/IRF4/MUM1(+), infrequently expresses BCL2 and is cytogenetically less complex [144]. Moreover, it does not show the impairment of TP53 and MYC signaling pathway typically observed in DLBCL or HGBCL with MYC and BCL2 rearrangements, whereas it exhibits impairment of E2F targets [148]. Because of its less distinctive biological features [141], HGBCL with MYC and BCL6 rearrangements, regarded as a provisional entity in ICC, is considered in the WHO-HAEM5 as a genetic subtypes of DLBCL, NOS and HGBCL, NOS, respectively.
HGBCL, NOS defines a rare molecularly heterogeneous subset of cases with blastoid or Burkitt-like cytology that do not have double-hit cytogenetics and do not readily fit within the categories of DLBCL, NOS or Burkitt lymphoma [149]. HGBCL, NOS usually shows a GCB phenotype and about half of cases carry a single-hit MYC rearrangement. Clinically, it occurs mostly in older adults (median age, 70 years) and it is characterized by high LDH, high IPI, extranodal involvement, CNS invasion and a more aggressive behavior than DLBCL [150]. Optimal treatment of HGBCL, NOS, remains uncertain because of the rarity of the tumor and the diagnostic variability. Patients presenting with aggressive disease should be treated with intensified Burkitt lymphoma-like regimens [150]. HGBCL also appears to respond well to immunotherapy with CAR-T cells [151].
In the WHO-HAEM4, cases with HGBCL (or DLBCL) morphology expressing TdT were classified as lymphoblastic leukemia/lymphoma. Now, based on mutational studies, CD34 negativity and presence of isolated or double-hit MYC rearrangement [152, 153], both WHO-HAEM5 and ICC recommend to consider these cases as DLBCL or HGBCL, NOS with “expression of TdT” (Fig. 4).

A Diffuse infiltration by large-size tumor cells (Hematoxylin-eosin; ×400) with evident nucleoli (inset; ×600). B The majority of the neoplastic cells strongly express TdT. C, D Tumor cells are CD79a negative but strongly express CD79b (TdT, CD79a, CD79b, Immunoperoxidase staining; ×400).
Burkitt lymphoma (BL)
The definition of BL remains unchanged in WHO-HAEM5 and ICC. BL is a very aggressive tumor characterized by a monotonous proliferation of medium-sized tumor cells, often associated with a “starry sky” pattern, GCB phenotype (CD10+, BCL6+, BCL2−), high proliferative index (Ki67 > 90%), IG::MYC juxtaposition and mutations involving TCF3 (E2A) or its repressor ID3 [154]. Three subtypes of BL are recognized in WHO-HAEM4: (1) “endemic”; (2) “non-endemic or sporadic”; and (3) “immunodeficiency-associated”. WHO-HAEM5 believes BL is better defined by molecular features than by epidemiologic criteria/geographic location [155,156,157,158]. Therefore, it emphasizes the distinction of BL in two subtypes: EBV-positive BL and EBV-negative BL, to reflect the dual mechanism of BL pathogenesis, i.e., virus-driven versus mutational-driven. EBV-positive cases exhibit higher levels of somatic hypermutation particularly in non-coding sequences close to the transcription start site [156] and harbor fewer driver mutations, including those affecting TCF3 and ID3 [156]. ICC recommends to classifying BL cases expressing TdT as B-lymphoblastic leukemia/lymphomas with MYC rearrangement rather than BL. In fact, they display phenotypic and molecular features of precursor B cells, including IG::MYC translocations arising from aberrant VDJ recombination, frequent lack of a productive IGH rearrangement and recurrent NRAS/KRAS mutations [159]. Separating these cases from BL is clinically important.
Hodgkin lymphoma
cHL is a GC-derived tumor characterized by a low number of neoplastic Hodgkin and Reed-Sternberg (H-RS) cells with a defective B-cell program that are immersed in an immunosuppressive microenvironment [160]. Genomic studies on microdissected tumor cells showed deregulation of the JAK-STAT pathway due to genetic alterations in STAT3, STAT5B, JAK1, JAK2, and PTPN1 in about 90% of cHL [161]. This finding supports the role of JAK-STAT pathway activation [161], among other genetic alterations [160], in cHL pathogenesis. In both ICC and WHO-HAEM5, diagnostic criteria for cHL remain unchanged. Immunohistochemical detection of CD30, CD15, IRF4/MUM1, PAX5, CD20, CD3 and LMP1 or EBER in situ hybridization is recommended. When H-RS cells are numerous and express CD20, cases should be investigated for the presence of additional B-cell markers, to exclude mediastinal gray zone lymphoma (MGZL). Moreover, cHL should be distinguished from other mimickers, e.g., lymphoproliferative disorders arising in the context of immune deficiency, that may contain EBV-positive H-RS-like cells [3, 4].
In ICC, the term nodular lymphocyte predominant Hodgkin lymphoma (NLPHL) has been changed into that of nodular lymphocyte predominant B-cell lymphoma. Conversely, WHO-HAEM5 felt the change of terminology was still premature and preferred to maintain the term of NLPHL, also to avoid interference with ongoing clinical trials [4]. The ICC decision was based on major biological and clinical differences between NLPHL and cHL, e.g., retention of functional B-cell program in NLPHL and close relationship of NLPHL to T-cell/histiocyte-rich large B-cell lymphoma [162, 163]. ICC also advises that “Fan patterns” A, B and C or Grade 1, should be distinguished from the less common “Fan patterns” D, E and F or Grade 2 [164]. The latter are usually associated with loss of nodularity, increased infiltration by T cells and a more aggressive clinical course. Cases with grade 2 histology may warrant treatment as DLBCL and patients with advanced stage NLPHL respond well to CHOP regimen [165]. Rare cases of NLPHL are EBV+ but the clinical significance of this finding remains unclear [166].
Mediastinal gray zone lymphoma (MGZL)
MGZL shows overlapping features with primary mediastinal B-cell lymphoma (PMBL) and cHL (especially nodular sclerosis). In both ICC and WHO-HAEM5, this entity now replaces the term “B-cell-lymphoma, unclassifiable with features intermediate between DLBCL and classic Hodgkin lymphoma” of WHO-HAEM4. Evidence that MGZL represents a true biological continuum with cHL and PMBL rather than a morphological mimic is supported by immunophenotypic patterns, gene expression profiles, methylation and mutational studies showing intermediate biological features between cHL and PMBL [167,168,169,170]. The concept of MGZL is further reinfocsed by the finding that nodular sclerosis cHL and PMBL developing sequentially may have common clonal origin [171]. These findings most likely reflect derivation of cHL, PMBL and MGZL (all located in the anterior mediastinum) from thymic B cells [172].
MGZL most frequently presents in young men (average 30 years) [173] with a bulky mediastinal mass and occasionally with supraclavicular lymphadenopathy and shows an inferior survival as compared to cHL, PMBL or DLBCL.The diagnosis of MGZL is based both on morphological criteria (e.g., high number of tumor cells) and immunohistological findings (expression of >1 B-cell marker in a significant percentage of neoplastic cells) [174] (Fig. 5). Cases with otherwise typical nodular sclerosis cHL that express CD20 but are negative for other B-cell markers should not be designed as MGZL [3]. MGZL is rarely EBV-positive [175]. The features of MGZL as compared to cHL and PMBL are shown in Table 2. Cases with morphologic and immunophenotypic features similar to MGZL, but occurring outside the mediastinum, show different gene expression profiles and genetic alterations [169, 176]. Thus, they should be classified as DLBCL, NOS.

A Diffuse proliferation of large tumor cells with wide clear cytoplasm (Hematoxylin and eosin; ×400); B Neoplastic cells strongly express CD30 (immunoperoxidase staining ×400). C Double staining showing a small percentage of reactive CD3-positive T cells (red) together with many surface CD19-positive tumor cells (brown); D Tumor cells from the same case are also double stained for MUM1/IRF4 (brown) and surface CD20 (red) (×400) (C, D Double immunoperoxidase staining using different enzyme substrates).
Multiple myeloma/plasma cell neoplasms
Non-IgM monoclonal gammopathy of undetermined significance (non-IgM MGUS) usually represents a precursor of MM [177]. Monoclonal gammopathy of renal or clinical significance (MGRS and MGCS) is characterized by a plasma cell or B-cell proliferation not meeting criteria for malignancy but secreting a monoclonal immunoglobulin or immunoglobulin fragment leading to kidney injury [178, 179]. The ICC describes MGRS/MGCS as a clinical feature of non-IgM MGUS whilst the WHO-HAEM5 regards it as an entity.
In the ICC classification [3], the term MM replaces that of “plasma cell myeloma” of WHO-HAEM4. The WHO-HAEM5 continues to use the term PCM. Based on progresses in cytogenetic/FISH studies, the ICC recognizes four mutually exclusive cytogenetic entities: (1) MM with CCND family translocations; (2) MM with NSD2 translocation; (3) MM with MAF family translocation; and (4) MM with hyperdiploidy. MM without cytogenetic abnormalities is defined as a separate entity named MM, NOS (Table 1). Such a distinction will likely facilitate the evaluation of response to new drugs, including immunomodulatory agents, proteasome inhibitors, monoclonal antibodies and CAR-T cells, according to cytogenetic features. These drugs have significantly improved the survival of patients with MM [180]. Translating mutational studies into classification was felt premature because of the frequent subclonal evolution and spatial genomic heterogeneity typical of MM/PCM [181,182,183,184].
The t(11;14), the most common cytogenetic abnormality (20–30%) in MM/PCM, leads to hyperexpression of cyclin D1 and correlates with increased sensitivity to venetoclax [185]. Translocation t(4;14) (12–15% of patients) [186] is specific to MM/PCM and deregulates the FGFR3 and NSD2 genes, the latter being responsible for poor prognosis [186]. Concomitant 1p32 deletion can significantly worsen the prognostic impact of t(4;14) [187]. Interestingly, the widespread use of bortezomib may have contributed to the reduction of the unfavorable prognosis of t(4;14) [188]. The translocation t(14;16) involving cMAF occurs at a much lower frequency (3.5%) [186] and its prognostic impact yet remains controversial [186, 189, 190]. Hyperdiploidy accounts for about 55% of cases [186]. The trisomies preferentially affect chromosomes 3, 5, 7, 9, 11, 15, 19, and 21. However, only trisomies 3 and 5 are associated with a better prognosis whilst trisomy 21 has an unfavorable impact [191]. The 17p deletion associates with high-risk, especially when the clone size is 55–60% by FISH [192] and the patients harbor a “double-hit” biallelic inactivation of TP53 [193].
Currently, MM/PCM patients receive similar treatment independently by the risk category. However, patients with high-risk disease still represent an unmet medical need with poor prognosis (death within the first 3 years from diagnosis) and should be identified for choosing the most efficient treatment strategy that maximizes the depth of response [194]. Inclusion of MM/PCM genetic categories in the classification may help to accelerate this process. The increasing importance of measurable residual disease [195] or PET/CT [196] in evaluating prognosis and risk stratification in MM/PCM is also recognized.
Smoldering/asymptomatic MM/PCM lacking features of active disease (SLiM CRAB criteria) [42] exhibits broad variability in progression to active MM/PCM. Patients suited for early therapeutic intervention are selected according to risk stratification models [197]. Solitary plasmacytomas of bone and primary extramedullary plasmacytomas have low-moderate risk for progression to MM/PCM [198]. Diagnosis, especially of solitary plasmacytomas of bone should be based on <10% clonal plasma cells detected by flow cytometry since this criterion is prognostically relevant [199].
Primary amyloidosis is termed immunoglobulin-related (AL) amyloidosis according to WHO-HAEM5 and Ig light chain (AL) amyloidosis according to ICC. The latter also emphasizes the importance of separating systemic AL amyloidosis from the localized form (Table 1), a rare disorder with excellent prognosis rarely progressing to systemic AL amyloidosis [200]. The WHO-HAEM4 recognized under the “plasma cell neoplasm with paraneoplastic syndrome” the POEMS and TEMPI syndromes as provisional entities. The TEMPI syndrome is characterized by telangiectasias, elevated erythropoietin, erythrocytosis, monoclonal gammopathy, perinephric fluid collection and intrapulmonary shunting [201]. Its diagnosis is mainly based upon clinical and imaging studies and it is now a defined entity in WHO-HAEM5. The AESOP syndrome is a newly recognized syndrome characterized by adenopathy with Castleman-like features and extensive skin patch overlying a plasmacytoma [202] that is regarded as an entity in WHO-HAEM5 but not in ICC.
Conclusions
The clinico-pathological, immunophenotypic and molecular informations gained during the past 5 years in the field of lymphoid neoplasms have contributed to refining the diagnostic criteria of several entities, to upgrade entities previously defined as provisional and to identify new entities. This is reflected in the changes reported in the WHO-HAEM5 and ICC classifications as compared to WHO-HAEM4. However, in several areas (e.g., DLBCL, NOS), incorporation of the molecular data into the new classifications was still regarded as premature. Our comparative report of the WHO-HAEM5 and ICC classifications of mature B-cell neoplasms may hopefully serve as a tool to facilitate the work of pathologists, hematologists and researchers involved in the diagnosis and treatment of lymphomas.
However, two additional comments may be worthy. First, after more than 20 years, we have once again two classifications. The hope is to come as soon as possible to a unifying approach since there is urgent need of a common language, which can be shared by the international community. This is in the interest of patients, clinicians and pathologists. Secondly, both the WHO-HAEM5 and ICC are based increasingly on molecular data, which implies the need for a network of reference centers, where expertise and facilities are available. In fact, the achievement of the correct diagnosis, the costs for the techniques applied may not be affordable at the community hospital level. The risk is that such a specialized approach may penalize developing countries. The latter should be supported on both economic and technical grounds. One way of achieving this goal could be to devote efforts for education [203], technology transfer and the production of new monoclonal antibodies against mutated epitopes that may serve as surrogates for molecular studies, as those we now use to diagnose some entities, such as NPM1-mutated AML [204] or ALK+ anaplastic large cell lymphoma [205].
References
Harris NL, Jaffe ES, Stein H, Banks PM, Chan JK, Cleary ML, et al. A revised European-American classification of lymphoid neoplasms: a proposal from the International Lymphoma Study Group. Blood. 1994;84:1361–92.
Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375–90.
Campo E, Jaffe ES, Cook JR, Quintanilla-Martinez L, Swerdlow SH, Anderson KC, et al. The International Consensus Classification of mature lymphoid neoplasms: a report from the Clinical Advisory Committee. Blood. 2022;140:1229–53.
Alaggio R, Amador C, Anagnostopoulos I, Attygalle AD, Araujo IBO, Berti E, et al. The 5th edition of the World Health Organization Classification of haematolymphoid tumours: lymphoid neoplasms. Leukemia. 2022;36:1720–48.
Rawstron AC, Kreuzer KA, Soosapilla A, Spacek M, Stehlikova O, Gambell P, et al. Reproducible diagnosis of chronic lymphocytic leukemia by flow cytometry: an European Research Initiative on CLL (ERIC) & European Society for Clinical Cell Analysis (ESCCA) Harmonisation project. Cytom B Clin Cytom. 2018;94:121–8.
Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Dohner H, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131:2745–60.
Rossi D, Khiabanian H, Spina V, Ciardullo C, Bruscaggin A, Fama R, et al. Clinical impact of small TP53 mutated subclones in chronic lymphocytic leukemia. Blood. 2014;123:2139–47.
Nadeu F, Delgado J, Royo C, Baumann T, Stankovic T, Pinyol M, et al. Clinical impact of clonal and subclonal TP53, SF3B1, BIRC3, NOTCH1, and ATM mutations in chronic lymphocytic leukemia. Blood. 2016;127:2122–30.
Bomben R, Rossi FM, Vit F, Bittolo T, D’Agaro T, Zucchetto A, et al. TP53 mutations with low variant allele frequency predict short survival in chronic lymphocytic leukemia. Clin Cancer Res. 2021;27:5566–75.
Nadeu F, Royo R, Clot G, Duran-Ferrer M, Navarro A, Martin S, et al. IGLV3-21R110 identifies an aggressive biological subtype of chronic lymphocytic leukemia with intermediate epigenetics. Blood. 2021;137:2935–46.
Hallek M, Al-Sawaf O. Chronic lymphocytic leukemia: 2022 update on diagnostic and therapeutic procedures. Am J Hematol. 2021;96:1679–705.
Baliakas P, Jeromin S, Iskas M, Puiggros A, Plevova K, Nguyen-Khac F, et al. Cytogenetic complexity in chronic lymphocytic leukemia: definitions, associations, and clinical impact. Blood. 2019;133:1205–16.
Fischer K, Al-Sawaf O, Bahlo J, Fink AM, Tandon M, Dixon M, et al. Venetoclax and obinutuzumab in patients with CLL and coexisting conditions. N Engl J Med. 2019;380:2225–36.
Gine E, Martinez A, Villamor N, Lopez-Guillermo A, Camos M, Martinez D, et al. Expanded and highly active proliferation centers identify a histological subtype of chronic lymphocytic leukemia (“accelerated” chronic lymphocytic leukemia) with aggressive clinical behavior. Haematologica. 2010;95:1526–33.
Barnea Slonim L, Ma S, Behdad A, Chen Q. Pseudo-Richter transformation of chronic lymphocytic leukaemia/small lymphocytic lymphoma following ibrutinib interruption: a diagnostic pitfall. Br J Haematol. 2020;191:e22–25.
Hampel PJ, Cherng HJ, Call TG, Ding W, Khanlari M, McPhail ED, et al. Incidental Richter transformation in chronic lymphocytic leukemia patients during temporary interruption of ibrutinib. Blood Adv. 2020;4:4508–11.
El Hussein S, Khoury JD, Medeiros LJ. B-prolymphocytic leukemia: is it time to retire this entity? Ann Diagn Pathol. 2021;54:151790.
Gordon MJ, Raess PW, Young K, Spurgeon SEF, Danilov AV. Ibrutinib is an effective treatment for B-cell prolymphocytic leukaemia. Br J Haematol. 2017;179:501–3.
Xing L, He Q, Xie L, Wang H, Li Z. Zanubrutinib, rituximab and lenalidomide induces deep and durable remission in TP53-mutated B-cell prolymphocytic leukemia: a case report and literature review. Haematologica. 2022;107:1226–8.
Patil N, Went RG. Venetoclax is an option in B-cell prolymphocytic leukaemia following progression on B-cell receptor pathway inhibitors. Br J Haematol. 2019;186:e80–2.
Rossi D, Bertoni F, Zucca E. Marginal-zone lymphomas. N Engl J Med. 2022;386:568–81.
Clipson A, Wang M, de Leval L, Ashton-Key M, Wotherspoon A, Vassiliou G, et al. KLF2 mutation is the most frequent somatic change in splenic marginal zone lymphoma and identifies a subset with distinct genotype. Leukemia. 2015;29:1177–85.
Campos-Martin Y, Martinez N, Martinez-Lopez A, Cereceda L, Casado F, Algara P, et al. Clinical and diagnostic relevance of NOTCH2-and KLF2-mutations in splenic marginal zone lymphoma. Haematologica. 2017;102:e310–12.
Rossi D, Trifonov V, Fangazio M, Bruscaggin A, Rasi S, Spina V, et al. The coding genome of splenic marginal zone lymphoma: activation of NOTCH2 and other pathways regulating marginal zone development. J Exp Med. 2012;209:1537–51.
Righi S, Novero D, Godio L, Bertuzzi C, Bacci F, Agostinelli C, et al. Myeloid nuclear differentiation antigen: an aid in differentiating lymphoplasmacytic lymphoma and splenic marginal zone lymphoma in bone marrow biopsies at presentation. Hum Pathol. 2022;124:67–75.
Angelova EA, Medeiros LJ, Wang W, Muzzafar T, Lu X, Khoury JD, et al. Clinicopathologic and molecular features in hairy cell leukemia-variant: single institutional experience. Mod Pathol. 2018;31:1717–32.
Waterfall JJ, Arons E, Walker RL, Pineda M, Roth L, Killian JK, et al. High prevalence of MAP2K1 mutations in variant and IGHV4-34-expressing hairy-cell leukemias. Nat Genet. 2014;46:8–10.
Tiacci E, Schiavoni G, Forconi F, Santi A, Trentin L, Ambrosetti A, et al. Simple genetic diagnosis of hairy cell leukemia by sensitive detection of the BRAF-V600E mutation. Blood. 2012;119:192–5.
Falini B, De Carolis L, Tiacci E. How I treat refractory/relapsed hairy cell leukemia with BRAF inhibitors. Blood. 2022;139:2294–305.
Kang C. Moxetumomab pasudotox in hairy cell leukaemia: a profile of its use. Clin Drug Investig. 2021;41:829–34.
Andritsos LA, Grieselhuber NR, Anghelina M, Rogers KA, Roychowdhury S, Reeser JW, et al. Trametinib for the treatment of IGHV4-34, MAP2K1-mutant variant hairy cell leukemia. Leuk Lymphoma. 2018;59:1008–11.
Favre R, Manzoni D, Traverse-Glehen A, Verney A, Jallades L, Callet-Bauchu E, et al. Usefulness of CD200 in the differential diagnosis of SDRPL, SMZL, and HCL. Int J Lab Hematol. 2018;40:e59–62.
Curiel-Olmo S, Mondejar R, Almaraz C, Mollejo M, Cereceda L, Mares R, et al. Splenic diffuse red pulp small B-cell lymphoma displays increased expression of cyclin D3 and recurrent CCND3 mutations. Blood. 2017;129:1042–5.
Jallades L, Baseggio L, Sujobert P, Huet S, Chabane K, Callet-Bauchu E, et al. Exome sequencing identifies recurrent BCOR alterations and the absence of KLF2, TNFAIP3 and MYD88 mutations in splenic diffuse red pulp small B-cell lymphoma. Haematologica. 2017;102:1758–66.
Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. MYD88 L265P somatic mutation in Waldenstrom’s macroglobulinemia. N Engl J Med. 2012;367:826–33.
Mori N, Ohwashi M, Yoshinaga K, Mitsuhashi K, Tanaka N, Teramura M, et al. L265P mutation of the MYD88 gene is frequent in Waldenstrom’s macroglobulinemia and its absence in myeloma. PLoS ONE. 2013;8:e80088.
Jimenez C, Sebastian E, Chillon MC, Giraldo P, Mariano Hernandez J, Escalante F, et al. MYD88 L265P is a marker highly characteristic of, but not restricted to, Waldenstrom’s macroglobulinemia. Leukemia. 2013;27:1722–8.
Treon SP, Gustine J, Xu L, Manning RJ, Tsakmaklis N, Demos M, et al. MYD88 wild-type Waldenstrom macroglobulinaemia: differential diagnosis, risk of histological transformation, and overall survival. Br J Haematol. 2018;180:374–80.
Hunter ZR, Xu L, Tsakmaklis N, Demos MG, Kofides A, Jimenez C, et al. Insights into the genomic landscape of MYD88 wild-type Waldenstrom macroglobulinemia. Blood Adv. 2018;2:2937–46.
Treon SP, Cao Y, Xu L, Yang G, Liu X, Hunter ZR. Somatic mutations in MYD88 and CXCR4 are determinants of clinical presentation and overall survival in Waldenstrom macroglobulinemia. Blood. 2014;123:2791–6.
Kaiser LM, Hunter ZR, Treon SP, Buske C. CXCR4 in Waldenstrom’s macroglobulinema: chances and challenges. Leukemia. 2021;35:333–45.
Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, Mateos MV, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15:e538–48.
Randen U, Troen G, Tierens A, Steen C, Warsame A, Beiske K, et al. Primary cold agglutinin-associated lymphoproliferative disease: a B-cell lymphoma of the bone marrow distinct from lymphoplasmacytic lymphoma. Haematologica. 2014;99:497–504.
Malecka A, Delabie J, Ostlie I, Tierens A, Randen U, Berentsen S, et al. Cold agglutinin-associated B-cell lymphoproliferative disease shows highly recurrent gains of chromosome 3 and 12 or 18. Blood Adv. 2020;4:993–6.
Malecka A, Troen G, Tierens A, Ostlie I, Malecki J, Randen U, et al. Frequent somatic mutations of KMT2D (MLL2) and CARD11 genes in primary cold agglutinin disease. Br J Haematol. 2018;183:838–42.
Rodriguez-Sevilla JJ, Salar A. Recent advances in the genetic of MALT lymphomas. Cancers. 2022;14:176.
Falini B, Tiacci E, Pucciarini A, Bigerna B, Kurth J, Hatzivassiliou G, et al. Expression of the IRTA1 receptor identifies intraepithelial and subepithelial marginal zone B cells of the mucosa-associated lymphoid tissue (MALT). Blood. 2003;102:3684–92.
Bob R, Falini B, Marafioti T, Paterson JC, Pileri S, Stein H. Nodal reactive and neoplastic proliferation of monocytoid and marginal zone B cells: an immunoarchitectural and molecular study highlighting the relevance of IRTA1 and T-bet as positive markers. Histopathology. 2013;63:482–98.
Falini B, Agostinelli C, Bigerna B, Pucciarini A, Pacini R, Tabarrini A, et al. IRTA1 is selectively expressed in nodal and extranodal marginal zone lymphomas. Histopathology. 2012;61:930–41.
Quintanilla-Martinez L, Sander B, Chan JK, Xerri L, Ott G, Campo E, et al. Indolent lymphomas in the pediatric population: follicular lymphoma, IRF4/MUM1+ lymphoma, nodal marginal zone lymphoma and chronic lymphocytic leukemia. Virchows Arch. 2016;468:141–57.
Edinger JT, Kant JA, Swerdlow SH. Cutaneous marginal zone lymphomas have distinctive features and include 2 subsets. Am J Surg Pathol. 2010;34:1830–41.
Brenner I, Roth S, Puppe B, Wobser M, Rosenwald A, Geissinger E. Primary cutaneous marginal zone lymphomas with plasmacytic differentiation show frequent IgG4 expression. Mod Pathol. 2013;26:1568–76.
Carlsen ED, Swerdlow SH, Cook JR, Gibson SE. Class-switched primary cutaneous marginal zone lymphomas are frequently IgG4-positive and have features distinct from IgM-positive cases. Am J Surg Pathol. 2019;43:1403–12.
van Maldegem F, van Dijk R, Wormhoudt TA, Kluin PM, Willemze R, Cerroni L, et al. The majority of cutaneous marginal zone B-cell lymphomas expresses class-switched immunoglobulins and develops in a T-helper type 2 inflammatory environment. Blood. 2008;112:3355–61.
Carlsen ED, Bhavsar S, Cook JR, Swerdlow SH. IRTA1 positivity helps identify a MALT-lymphoma-like subset of primary cutaneous marginal zone lymphomas, largely but not exclusively defined by IgM expression. J Cutan Pathol. 2022;49:55–60.
Maurus K, Appenzeller S, Roth S, Kuper J, Rost S, Meierjohann S, et al. Panel sequencing shows recurrent genetic FAS alterations in primary cutaneous marginal zone lymphoma. J Invest Dermatol. 2018;138:1573–81.
Laurent C, Adelaide J, Guille A, Tesson B, Gat E, Evrard S, et al. High-grade follicular lymphomas exhibit clinicopathologic, cytogenetic, and molecular diversity extending beyond grades 3A and 3B. Am J Surg Pathol. 2021;45:1324–36.
Salaverria I, Philipp C, Oschlies I, Kohler CW, Kreuz M, Szczepanowski M, et al. Translocations activating IRF4 identify a subtype of germinal center-derived B-cell lymphoma affecting predominantly children and young adults. Blood. 2011;118:139–47.
Siddiqi IN, Friedman J, Barry-Holson KQ, Ma C, Thodima V, Kang I, et al. Characterization of a variant of t(14;18) negative nodal diffuse follicular lymphoma with CD23 expression, 1p36/TNFRSF14 abnormalities, and STAT6 mutations. Mod Pathol. 2016;29:570–81.
Shah NN, Szabo A, Saba R, Strelec L, Kodali D, Vaughn JL, et al. Multicenter analysis of advanced stage grade 3A follicular lymphoma outcomes by frontline treatment regimen. Clin Lymphoma Myeloma Leuk. 2019;19:95–102.
Barraclough A, Bishton M, Cheah CY, Villa D, Hawkes EA. The diagnostic and therapeutic challenges of grade 3B follicular lymphoma. Br J Haematol. 2021;195:15–24.
Koch K, Richter J, Hanel C, Huttmann A, Duhrsen U, Klapper W. Follicular lymphoma grade 3B and diffuse large B-cell lymphoma present a histopathological and molecular continuum lacking features of progression/transformation. Haematologica. 2022;107:2144–53.
Metter GE, Nathwani BN, Burke JS, Winberg CD, Mann RB, Barcos M, et al. Morphological subclassification of follicular lymphoma: variability of diagnoses among hematopathologists, a collaborative study between the Repository Center and Pathology Panel for Lymphoma Clinical Studies. J Clin Oncol. 1985;3:25–38.
Rimsza LM, Li H, Braziel RM, Spier CM, Persky DO, Dunlap J, et al. Impact of histological grading on survival in the SWOG S0016 follicular lymphoma cohort. Haematologica. 2018;103:e151–3.
Liu Q, Salaverria I, Pittaluga S, Jegalian AG, Xi L, Siebert R, et al. Follicular lymphomas in children and young adults: a comparison of the pediatric variant with usual follicular lymphoma. Am J Surg Pathol. 2013;37:333–43.
Louissaint A Jr., Schafernak KT, Geyer JT, Kovach AE, Ghandi M, Gratzinger D, et al. Pediatric-type nodal follicular lymphoma: a biologically distinct lymphoma with frequent MAPK pathway mutations. Blood. 2016;128:1093–100.
Schmidt J, Ramis-Zaldivar JE, Nadeu F, Gonzalez-Farre B, Navarro A, Egan C, et al. Mutations of MAP2K1 are frequent in pediatric-type follicular lymphoma and result in ERK pathway activation. Blood. 2017;130:323–7.
Finn LS, Viswanatha DS, Belasco JB, Snyder H, Huebner D, Sorbara L, et al. Primary follicular lymphoma of the testis in childhood. Cancer. 1999;85:1626–35.
Lones MA, Raphael M, McCarthy K, Wotherspoon A, Terrier-Lacombe MJ, Ramsay AD, et al. Primary follicular lymphoma of the testis in children and adolescents. J Pediatr Hematol Oncol. 2012;34:68–71.
Katzenberger T, Kalla J, Leich E, Stocklein H, Hartmann E, Barnickel S, et al. A distinctive subtype of t(14;18)-negative nodal follicular non-Hodgkin lymphoma characterized by a predominantly diffuse growth pattern and deletions in the chromosomal region 1p36. Blood. 2009;113:1053–61.
Nann D, Ramis-Zaldivar JE, Muller I, Gonzalez-Farre B, Schmidt J, Egan C, et al. Follicular lymphoma t(14;18)-negative is genetically a heterogeneous disease. Blood Adv. 2020;4:5652–65.
Morschhauser F, Tilly H, Chaidos A, McKay P, Phillips T, Assouline S, et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: an open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol. 2020;21:1433–42.
Pastore A, Jurinovic V, Kridel R, Hoster E, Staiger AM, Szczepanowski M, et al. Integration of gene mutations in risk prognostication for patients receiving first-line immunochemotherapy for follicular lymphoma: a retrospective analysis of a prospective clinical trial and validation in a population-based registry. Lancet Oncol. 2015;16:1111–22.
Jacobson CA, Chavez JC, Sehgal AR, William BM, Munoz J, Salles G, et al. Axicabtagene ciloleucel in relapsed or refractory indolent non-Hodgkin lymphoma (ZUMA-5): a single-arm, multicentre, phase 2 trial. Lancet Oncol. 2022;23:91–103.
Wlodarska I, Dierickx D, Vanhentenrijk V, Van Roosbroeck K, Pospisilova H, Minnei F, et al. Translocations targeting CCND2, CCND3, and MYCN do occur in t(11;14)-negative mantle cell lymphomas. Blood. 2008;111:5683–90.
Salaverria I, Royo C, Carvajal-Cuenca A, Clot G, Navarro A, Valera A, et al. CCND2 rearrangements are the most frequent genetic events in cyclin D1(-) mantle cell lymphoma. Blood. 2013;121:1394–402.
Fu K, Weisenburger DD, Greiner TC, Dave S, Wright G, Rosenwald A, et al. Cyclin D1-negative mantle cell lymphoma: a clinicopathologic study based on gene expression profiling. Blood. 2005;106:4315–21.
Martin-Garcia D, Navarro A, Valdes-Mas R, Clot G, Gutierrez-Abril J, Prieto M, et al. CCND2 and CCND3 hijack immunoglobulin light-chain enhancers in cyclin D1(-) mantle cell lymphoma. Blood. 2019;133:940–51.
Aukema SM, Croci GA, Bens S, Oehl-Huber K, Wagener R, Ott G, et al. Mantle cell lymphomas with concomitant MYC and CCND1 breakpoints are recurrently TdT positive and frequently show high-grade pathological and genetic features. Virchows Arch. 2021;479:133–45.
Hoster E, Rosenwald A, Berger F, Bernd HW, Hartmann S, Loddenkemper C, et al. Prognostic value of Ki-67 index, cytology, and growth pattern in mantle-cell lymphoma: results from randomized trials of the European Mantle Cell Lymphoma Network. J Clin Oncol. 2016;34:1386–94.
Armitage JO, Longo DL. Mantle-cell lymphoma. N Engl J Med. 2022;386:2495–506.
Tam CS, Anderson MA, Pott C, Agarwal R, Handunnetti S, Hicks RJ, et al. Ibrutinib plus venetoclax for the treatment of mantle-cell lymphoma. N Engl J Med. 2018;378:1211–23.
Wang ML, Jurczak W, Jerkeman M, Trotman J, Zinzani PL, Belada D, et al. Ibrutinib plus bendamustine and rituximab in untreated mantle-cell lymphoma. N Engl J Med. 2022;386:2482–94.
Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2020;382:1331–42.
Kumar A, Eyre TA, Lewis KL, Thompson MC, Cheah CY. New directions for mantle cell lymphoma in 2022. Am Soc Clin Oncol Educ Book. 2022;42:1–15.
Thompson ER, Nguyen T, Kankanige Y, Markham JF, Anderson MA, Handunnetti SM, et al. Single-cell sequencing demonstrates complex resistance landscape in CLL and MCL treated with BTK and BCL2 inhibitors. Blood Adv. 2022;6:503–8.
Nadeu F, Martin-Garcia D, Clot G, Diaz-Navarro A, Duran-Ferrer M, Navarro A, et al. Genomic and epigenomic insights into the origin, pathogenesis, and clinical behavior of mantle cell lymphoma subtypes. Blood. 2020;136:1419–32.
Royo C, Navarro A, Clot G, Salaverria I, Gine E, Jares P, et al. Non-nodal type of mantle cell lymphoma is a specific biological and clinical subgroup of the disease. Leukemia. 2012;26:1895–8.
Navarro A, Clot G, Royo C, Jares P, Hadzidimitriou A, Agathangelidis A, et al. Molecular subsets of mantle cell lymphoma defined by the IGHV mutational status and SOX11 expression have distinct biologic and clinical features. Cancer Res. 2012;72:5307–16.
Xu J, Wang L, Li J, Saksena A, Wang SA, Shen J, et al. SOX11-negative mantle cell lymphoma: clinicopathologic and prognostic features of 75 patients. Am J Surg Pathol. 2019;43:710–6.
Orchard J, Garand R, Davis Z, Babbage G, Sahota S, Matutes E, et al. A subset of t(11;14) lymphoma with mantle cell features displays mutated IgVH genes and includes patients with good prognosis, nonnodal disease. Blood. 2003;101:4975–81.
Durani U, Ansell SM. CD5+ diffuse large B-cell lymphoma: a narrative review. Leuk Lymphoma. 2021;62:3078–86.
Meriranta L, Pasanen A, Alkodsi A, Haukka J, Karjalainen-Lindsberg ML, Leppa S. Molecular background delineates outcome of double protein expressor diffuse large B-cell lymphoma. Blood Adv. 2020;4:3742–53.
Staiger AM, Ziepert M, Horn H, Scott DW, Barth TFE, Bernd HW, et al. Clinical impact of the cell-of-origin classification and the MYC/ BCL2 dual expressor status in diffuse large B-cell lymphoma treated within prospective clinical trials of the German High-Grade Non-Hodgkin’s Lymphoma Study Group. J Clin Oncol. 2017;35:2515–26.
Pasqualucci L, Dalla-Favera R. The genetic landscape of diffuse large B-cell lymphoma. Semin Hematol. 2015;52:67–76.
Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med. 2002;346:1937–47.
Falini B, Fizzotti M, Pucciarini A, Bigerna B, Marafioti T, Gambacorta M, et al. A monoclonal antibody (MUM1p) detects expression of the MUM1/IRF4 protein in a subset of germinal center B cells, plasma cells, and activated T cells. Blood. 2000;95:2084–92.
Hans CP, Weisenburger DD, Greiner TC, Gascoyne RD, Delabie J, Ott G, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood. 2004;103:275–82.
Younes A, Sehn LH, Johnson P, Zinzani PL, Hong X, Zhu J, et al. Randomized phase III trial of ibrutinib and rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in non-germinal center B-cell diffuse large B-cell lymphoma. J Clin Oncol. 2019;37:1285–95.
Leonard JP, Kolibaba KS, Reeves JA, Tulpule A, Flinn IW, Kolevska T, et al. Randomized phase II study of R-CHOP with or without bortezomib in previously untreated patients with non-germinal center B-cell-like diffuse large B-cell lymphoma. J Clin Oncol. 2017;35:3538–46.
Nowakowski GS, Hong F, Scott DW, Macon WR, King RL, Habermann TM, et al. Addition of lenalidomide to R-CHOP improves outcomes in newly diagnosed diffuse large B-cell lymphoma in a randomized phase II US Intergroup Study ECOG-ACRIN E1412. J Clin Oncol. 2021;39:1329–38.
Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. N Engl J Med. 2018;378:1396–407.
Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med. 2018;24:679–90.
Wright GW, Huang DW, Phelan JD, Coulibaly ZA, Roulland S, Young RM, et al. A Probabilistic classification tool for genetic subtypes of diffuse large B cell lymphoma with therapeutic implications. Cancer Cell. 2020;37:551–68.e514.
Morin RD, Arthur SE, Hodson DJ. Molecular profiling in diffuse large B-cell lymphoma: why so many types of subtypes? Br J Haematol. 2022;196:814–29.
Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2019;380:45–56.
King RL, Goodlad JR, Calaminici M, Dotlic S, Montes-Moreno S, Oschlies I, et al. Lymphomas arising in immune-privileged sites: insights into biology, diagnosis, and pathogenesis. Virchows Arch. 2020;476:647–65.
Kraan W, van Keimpema M, Horlings HM, Schilder-Tol EJ, Oud ME, Noorduyn LA, et al. High prevalence of oncogenic MYD88 and CD79B mutations in primary testicular diffuse large B-cell lymphoma. Leukemia. 2014;28:719–20.
Nakamura T, Tateishi K, Niwa T, Matsushita Y, Tamura K, Kinoshita M, et al. Recurrent mutations of CD79B and MYD88 are the hallmark of primary central nervous system lymphomas. Neuropathol Appl Neurobiol. 2016;42:279–90.
Pollari M, Bruck O, Pellinen T, Vahamurto P, Karjalainen-Lindsberg ML, Mannisto S, et al. PD-L1(+) tumor-associated macrophages and PD-1(+) tumor-infiltrating lymphocytes predict survival in primary testicular lymphoma. Haematologica. 2018;103:1908–14.
Booman M, Douwes J, Glas AM, Riemersma SA, Jordanova ES, Kok K, et al. Mechanisms and effects of loss of human leukocyte antigen class II expression in immune-privileged site-associated B-cell lymphoma. Clin Cancer Res. 2006;12:2698–705.
Mital P, Hinton BT, Dufour JM. The blood-testis and blood-epididymis barriers are more than just their tight junctions. Biol Reprod. 2011;84:851–8.
Frigault MJ, Dietrich J, Gallagher K, Roschewski M, Jordan JT, Forst D, et al. Safety and efficacy of tisagenlecleucel in primary CNS lymphoma: a phase 1/2 clinical trial. Blood. 2022;139:2306–15.
Frigault MJ, Dietrich J, Martinez-Lage M, Leick M, Choi BD, DeFilipp Z, et al. Tisagenlecleucel CAR T-cell therapy in secondary CNS lymphoma. Blood. 2019;134:860–6.
Fonseca R, Habermann TM, Colgan JP, O’Neill BP, White WL, Witzig TE, et al. Testicular lymphoma is associated with a high incidence of extranodal recurrence. Cancer. 2000;88:154–61.
Pham-Ledard A, Beylot-Barry M, Barbe C, Leduc M, Petrella T, Vergier B, et al. High frequency and clinical prognostic value of MYD88 L265P mutation in primary cutaneous diffuse large B-cell lymphoma, leg-type. JAMA Dermatol. 2014;150:1173–9.
Taniguchi K, Takata K, Chuang SS, Miyata-Takata T, Sato Y, Satou A, et al. Frequent MYD88 L265P and CD79B mutations in primary breast diffuse large B-cell lymphoma. Am J Surg Pathol. 2016;40:324–34.
Schrader AMR, Jansen PM, Willemze R, Vermeer MH, Cleton-Jansen AM, Somers SF, et al. High prevalence of MYD88 and CD79B mutations in intravascular large B-cell lymphoma. Blood. 2018;131:2086–9.
Salaverria I, Martin-Guerrero I, Wagener R, Kreuz M, Kohler CW, Richter J, et al. A recurrent 11q aberration pattern characterizes a subset of MYC-negative high-grade B-cell lymphomas resembling Burkitt lymphoma. Blood. 2014;123:1187–98.
Ferreiro JF, Morscio J, Dierickx D, Marcelis L, Verhoef G, Vandenberghe P, et al. Post-transplant molecularly defined Burkitt lymphomas are frequently MYC-negative and characterized by the 11q-gain/loss pattern. Haematologica. 2015;100:e275–9.
Rymkiewicz G, Grygalewicz B, Chechlinska M, Blachnio K, Bystydzienski Z, Romejko-Jarosinska J, et al. A comprehensive flow-cytometry-based immunophenotypic characterization of Burkitt-like lymphoma with 11q aberration. Mod Pathol. 2018;31:732–43.
Wagener R, Seufert J, Raimondi F, Bens S, Kleinheinz K, Nagel I, et al. The mutational landscape of Burkitt-like lymphoma with 11q aberration is distinct from that of Burkitt lymphoma. Blood. 2019;133:962–6.
Gonzalez-Farre B, Ramis-Zaldivar JE, Salmeron-Villalobos J, Balague O, Celis V, Verdu-Amoros J, et al. Burkitt-like lymphoma with 11q aberration: a germinal center-derived lymphoma genetically unrelated to Burkitt lymphoma. Haematologica. 2019;104:1822–9.
Horn H, Kalmbach S, Wagener R, Staiger AM, Huttl K, Mottok A, et al. A diagnostic approach to the identification of Burkitt-like lymphoma with 11q aberration in aggressive B-cell lymphomas. Am J Surg Pathol. 2021;45:356–64.
Falini B, Lazzi S. Pediatric large B-cell lymphoma with 11q aberration. Blood. 2022 (in press).
Ramis-Zaldivar JE, Gonzalez-Farre B, Balague O, Celis V, Nadeu F, Salmeron-Villalobos J, et al. Distinct molecular profile of IRF4-rearranged large B-cell lymphoma. Blood. 2020;135:274–86.
Natkunam Y, Goodlad JR, Chadburn A, de Jong D, Gratzinger D, Chan JK, et al. EBV-positive B-cell proliferations of varied malignant potential: 2015 SH/EAHP Workshop Report-Part 1. Am J Clin Pathol. 2017;147:129–52.
Natkunam Y, Gratzinger D, Chadburn A, Goodlad JR, Chan JKC, Said J, et al. Immunodeficiency-associated lymphoproliferative disorders: time for reappraisal? Blood. 2018;132:1871–8.
Nicolae A, Pittaluga S, Abdullah S, Steinberg SM, Pham TA, Davies-Hill T, et al. EBV-positive large B-cell lymphomas in young patients: a nodal lymphoma with evidence for a tolerogenic immune environment. Blood. 2015;126:863–72.
Bourbon E, Maucort-Boulch D, Fontaine J, Mauduit C, Sesques P, Safar V, et al. Clinicopathological features and survival in EBV-positive diffuse large B-cell lymphoma not otherwise specified. Blood Adv. 2021;5:3227–39.
Falini B, Lazzi S. Epstein-Barr virus-positive mucocutaneous ulcer of the stomach. Blood. 2022;140:1743.
Dojcinov SD, Venkataraman G, Raffeld M, Pittaluga S, Jaffe ES. EBV positive mucocutaneous ulcer—a study of 26 cases associated with various sources of immunosuppression. Am J Surg Pathol. 2010;34:405–17.
Ikeda T, Gion Y, Sakamoto M, Tachibana T, Nishikori A, Nishimura MF, et al. Clinicopathological analysis of 34 Japanese patients with EBV-positive mucocutaneous ulcer. Mod Pathol. 2020;33:2437–48.
Ikeda T, Gion Y, Nishimura Y, Nishimura MF, Yoshino T, Sato Y. Epstein-Barr virus-positive mucocutaneous ulcer: a unique and curious disease entity. Int J Mol Sci. 2021;22:1053.
Ishikawa E, Satou A, Nakamura M, Nakamura S, Fujishiro M. Epstein-Barr virus positive B-cell lymphoproliferative disorder of the gastrointestinal tract. Cancers. 2021;13:3815.
Yamakawa N, Fujimoto M, Kawabata D, Terao C, Nishikori M, Nakashima R, et al. A clinical, pathological, and genetic characterization of methotrexate-associated lymphoproliferative disorders. J Rheumatol. 2014;41:293–9.
Hart M, Thakral B, Yohe S, Balfour HH Jr., Singh C, Spears M, et al. EBV-positive mucocutaneous ulcer in organ transplant recipients: a localized indolent posttransplant lymphoproliferative disorder. Am J Surg Pathol. 2014;38:1522–9.
Alexanian S, Said J, Lones M, Pullarkat ST. KSHV/HHV8-negative effusion-based lymphoma, a distinct entity associated with fluid overload states. Am J Surg Pathol. 2013;37:241–9.
Kubota T, Sasaki Y, Shiozawa E, Takimoto M, Hishima T, Chong JM. Age and CD20 expression are significant prognostic factors in human Herpes Virus-8-negative effusion-based lymphoma. Am J Surg Pathol. 2018;42:1607–16.
Kaji D, Ota Y, Sato Y, Nagafuji K, Ueda Y, Okamoto M, et al. Primary human herpesvirus 8-negative effusion-based lymphoma: a large B-cell lymphoma with favorable prognosis. Blood Adv. 2020;4:4442–50.
Cucco F, Barrans S, Sha C, Clipson A, Crouch S, Dobson R, et al. Distinct genetic changes reveal evolutionary history and heterogeneous molecular grade of DLBCL with MYC/BCL2 double-hit. Leukemia. 2020;34:1329–41.
Ennishi D, Jiang A, Boyle M, Collinge B, Grande BM, Ben-Neriah S, et al. Double-hit gene expression signature defines a distinct subgroup of germinal center B-cell-like diffuse large B-cell lymphoma. J Clin Oncol. 2019;37:190–201.
Evrard SM, Pericart S, Grand D, Amara N, Escudie F, Gilhodes J, et al. Targeted next generation sequencing reveals high mutation frequency of CREBBP, BCL2 and KMT2D in high-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements. Haematologica. 2019;104:e154–7.
Pillai RK, Sathanoori M, Van Oss SB, Swerdlow SH. Double-hit B-cell lymphomas with BCL6 and MYC translocations are aggressive, frequently extranodal lymphomas distinct from BCL2 double-hit B-cell lymphomas. Am J Surg Pathol. 2013;37:323–32.
Rosenwald A, Bens S, Advani R, Barrans S, Copie-Bergman C, Elsensohn MH, et al. Prognostic significance of MYC rearrangement and translocation partner in diffuse large B-cell lymphoma: a study by the Lunenburg Lymphoma Biomarker Consortium. J Clin Oncol. 2019;37:3359–68.
Li S, Seegmiller AC, Lin P, Wang XJ, Miranda RN, Bhagavathi S, et al. B-cell lymphomas with concurrent MYC and BCL2 abnormalities other than translocations behave similarly to MYC/BCL2 double-hit lymphomas. Mod Pathol. 2015;28:208–17.
Li S, Desai P, Lin P, Yin CC, Tang G, Wang XJ, et al. MYC/BCL6 double-hit lymphoma (DHL): a tumour associated with an aggressive clinical course and poor prognosis. Histopathology. 2016;68:1090–8.
Kunstner A, Witte HM, Riedl J, Bernard V, Stolting S, Merz H, et al. Mutational landscape of high-grade B-cell lymphoma with MYC-, BCL2 and/or BCL6 rearrangements characterized by whole-exome sequencing. Haematologica. 2022;107:1850–63.
Huttl KS, Staiger AM, Richter J, Ott MM, Kalmbach S, Klapper W, et al. The “Burkitt-like” immunophenotype and genotype is rarely encountered in diffuse large B cell lymphoma and high-grade B cell lymphoma, NOS. Virchows Arch. 2021;479:575–83.
Olszewski AJ, Kurt H, Evens AM. Defining and treating high-grade B-cell lymphoma, NOS. Blood. 2022;140:943–54.
Jacobson CA, Hunter BD, Redd R, Rodig SJ, Chen PH, Wright K, et al. Axicabtagene ciloleucel in the non-trial setting: outcomes and correlates of response, resistance, and toxicity. J Clin Oncol. 2020;38:3095–106.
Khanlari M, Medeiros LJ, Lin P, Xu J, You MJ, Tang G, et al. Blastoid high-grade B-cell lymphoma initially presenting in bone marrow: a diagnostic challenge. Mod Pathol. 2022;35:419–26.
Bhavsar S, Liu YC, Gibson SE, Moore EM, Swerdlow SH. Mutational landscape of TdT+ large B-cell lymphomas supports their distinction from B-lymphoblastic neoplasms: a multiparameter study of a rare and aggressive entity. Am J Surg Pathol. 2022;46:71–82.
Schmitz R, Young RM, Ceribelli M, Jhavar S, Xiao W, Zhang M, et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature. 2012;490:116–20.
Kaymaz Y, Oduor CI, Yu H, Otieno JA, Ong’echa JM, Moormann AM, et al. Comprehensive transcriptome and mutational profiling of endemic Burkitt lymphoma reveals EBV type-specific differences. Mol Cancer Res. 2017;15:563–76.
Grande BM, Gerhard DS, Jiang A, Griner NB, Abramson JS, Alexander TB, et al. Genome-wide discovery of somatic coding and noncoding mutations in pediatric endemic and sporadic Burkitt lymphoma. Blood. 2019;133:1313–24.
Richter J, John K, Staiger AM, Rosenwald A, Kurz K, Michgehl U, et al. Epstein-Barr virus status of sporadic Burkitt lymphoma is associated with patient age and mutational features. Br J Haematol. 2022;196:681–9.
Panea RI, Love CL, Shingleton JR, Reddy A, Bailey JA, Moormann AM, et al. The whole-genome landscape of Burkitt lymphoma subtypes. Blood. 2019;134:1598–607.
Wagener R, Lopez C, Kleinheinz K, Bausinger J, Aukema SM, Nagel I, et al. IG-MYC (+) neoplasms with precursor B-cell phenotype are molecularly distinct from Burkitt lymphomas. Blood. 2018;132:2280–5.
Weniger MA, Kuppers R. Molecular biology of Hodgkin lymphoma. Leukemia. 2021;35:968–81.
Tiacci E, Ladewig E, Schiavoni G, Penson A, Fortini E, Pettirossi V, et al. Pervasive mutations of JAK-STAT pathway genes in classical Hodgkin lymphoma. Blood. 2018;131:2454–65.
Fan Z, Natkunam Y, Bair E, Tibshirani R, Warnke RA. Characterization of variant patterns of nodular lymphocyte predominant Hodgkin lymphoma with immunohistologic and clinical correlation. Am J Surg Pathol. 2003;27:1346–56.
Hartmann S, Doring C, Vucic E, Chan FC, Ennishi D, Tousseyn T, et al. Array comparative genomic hybridization reveals similarities between nodular lymphocyte predominant Hodgkin lymphoma and T cell/histiocyte rich large B cell lymphoma. Br J Haematol. 2015;169:415–22.
Hartmann S, Eichenauer DA, Plutschow A, Mottok A, Bob R, Koch K, et al. The prognostic impact of variant histology in nodular lymphocyte-predominant Hodgkin lymphoma: a report from the German Hodgkin Study Group (GHSG). Blood. 2013;122:4246–52.
Fanale MA, Cheah CY, Rich A, Medeiros LJ, Lai CM, Oki Y, et al. Encouraging activity for R-CHOP in advanced stage nodular lymphocyte-predominant Hodgkin lymphoma. Blood. 2017;130:472–7.
Huppmann AR, Nicolae A, Slack GW, Pittaluga S, Davies-Hill T, Ferry JA, et al. EBV may be expressed in the LP cells of nodular lymphocyte-predominant Hodgkin lymphoma (NLPHL) in both children and adults. Am J Surg Pathol. 2014;38:316–24.
Pittaluga S, Nicolae A, Wright GW, Melani C, Roschewski M, Steinberg S, et al. Gene expression profiling of mediastinal gray zone lymphoma and its relationship to primary mediastinal B-cell lymphoma and classical Hodgkin lymphoma. Blood Cancer Discov. 2020;1:155–61.
Sarkozy C, Chong L, Takata K, Chavez EA, Miyata-Takata T, Duns G, et al. Gene expression profiling of gray zone lymphoma. Blood Adv. 2020;4:2523–35.
Sarkozy C, Hung SS, Chavez EA, Duns G, Takata K, Chong LC, et al. Mutational landscape of gray zone lymphoma. Blood. 2021;137:1765–76.
Eberle FC, Rodriguez-Canales J, Wei L, Hanson JC, Killian JK, Sun HW, et al. Methylation profiling of mediastinal gray zone lymphoma reveals a distinctive signature with elements shared by classical Hodgkin’s lymphoma and primary mediastinal large B-cell lymphoma. Haematologica. 2011;96:558–66.
Singh K, Lezama LS, Kurzer J, Oak J, Schultz LM, Walkush A, et al. Targeted mutational profiling reveals clonal relationships in metachronous occurrence of classic Hodgkin and mediastinal large B-cell lymphomas. Am J Surg Pathol. 2022. Epub ahead of print.
Bosch-Schips J, Granai M, Quintanilla-Martinez L, Fend F. The grey zones of classic Hodgkin lymphoma. Cancers. 2022;14:742.
Oschlies I, Burkhardt B, Salaverria I, Rosenwald A, d’Amore ES, Szczepanowski M, et al. Clinical, pathological and genetic features of primary mediastinal large B-cell lymphomas and mediastinal gray zone lymphomas in children. Haematologica. 2011;96:262–8.
Wilson WH, Pittaluga S, Nicolae A, Camphausen K, Shovlin M, Steinberg SM, et al. A prospective study of mediastinal gray-zone lymphoma. Blood. 2014;124:1563–9.
Elsayed AA, Satou A, Eladl AE, Kato S, Nakamura S, Asano N. Grey zone lymphoma with features intermediate between diffuse large B-cell lymphoma and classical Hodgkin lymphoma: a clinicopathological study of 14 Epstein-Barr virus-positive cases. Histopathology. 2017;70:579–94.
Campo E, Jaffe ES. Taking gray zone lymphomas out of the shadows. Blood. 2021;137:1703–4.
Weiss BM, Abadie J, Verma P, Howard RS, Kuehl WM. A monoclonal gammopathy precedes multiple myeloma in most patients. Blood. 2009;113:5418–22.
Leung N, Bridoux F, Batuman V, Chaidos A, Cockwell P, D’Agati VD, et al. The evaluation of monoclonal gammopathy of renal significance: a consensus report of the International Kidney and Monoclonal Gammopathy Research Group. Nat Rev Nephrol. 2019;15:45–59.
Leung N, Bridoux F, Nasr SH. Monoclonal gammopathy of renal significance. N Engl J Med. 2021;384:1931–41.
Usmani SZ, Hoering A, Cavo M, Miguel JS, Goldschimdt H, Hajek R, et al. Clinical predictors of long-term survival in newly diagnosed transplant eligible multiple myeloma—an IMWG Research Project. Blood Cancer J. 2018;8:123.
Walker BA, Mavrommatis K, Wardell CP, Ashby TC, Bauer M, Davies FE, et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood. 2018;132:587–97.
Rasche L, Chavan SS, Stephens OW, Patel PH, Tytarenko R, Ashby C, et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nat Commun. 2017;8:268.
Manier S, Salem KZ, Park J, Landau DA, Getz G, Ghobrial IM. Genomic complexity of multiple myeloma and its clinical implications. Nat Rev Clin Oncol. 2017;14:100–13.
Boyle EM, Deshpande S, Tytarenko R, Ashby C, Wang Y, Bauer MA, et al. The molecular make up of smoldering myeloma highlights the evolutionary pathways leading to multiple myeloma. Nat Commun. 2021;12:293.
Kumar S, Kaufman JL, Gasparetto C, Mikhael J, Vij R, Pegourie B, et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood. 2017;130:2401–9.
Corre J, Munshi NC, Avet-Loiseau H. Risk factors in multiple myeloma: is it time for a revision? Blood. 2021;137:16–19.
Hebraud B, Magrangeas F, Cleynen A, Lauwers-Cances V, Chretien ML, Hulin C, et al. Role of additional chromosomal changes in the prognostic value of t(4;14) and del(17p) in multiple myeloma: the IFM experience. Blood. 2015;125:2095–100.
Avet-Loiseau H, Leleu X, Roussel M, Moreau P, Guerin-Charbonnel C, Caillot D, et al. Bortezomib plus dexamethasone induction improves outcome of patients with t(4;14) myeloma but not outcome of patients with del(17p). J Clin Oncol. 2010;28:4630–4.
Avet-Loiseau H, Malard F, Campion L, Magrangeas F, Sebban C, Lioure B, et al. Translocation t(14;16) and multiple myeloma: is it really an independent prognostic factor? Blood. 2011;117:2009–11.
Goldman-Mazur S, Jurczyszyn A, Castillo JJ, Waszczuk-Gajda A, Grzasko N, Radocha J, et al. A multicenter retrospective study of 223 patients with t(14;16) in multiple myeloma. Am J Hematol. 2020;95:503–9.
Chretien ML, Corre J, Lauwers-Cances V, Magrangeas F, Cleynen A, Yon E, et al. Understanding the role of hyperdiploidy in myeloma prognosis: which trisomies really matter? Blood. 2015;126:2713–9.
Thakurta A, Ortiz M, Blecua P, Towfic F, Corre J, Serbina NV, et al. High subclonal fraction of 17p deletion is associated with poor prognosis in multiple myeloma. Blood. 2019;133:1217–21.
Walker BA, Mavrommatis K, Wardell CP, Ashby TC, Bauer M, Davies F, et al. A high-risk, double-hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia. 2019;33:159–70.
Goldman-Mazur S, Kumar SK. Current approaches to management of high-risk multiple myeloma. Am J Hematol. 2021;96:854–71.
Cavo M, San-Miguel J, Usmani SZ, Weisel K, Dimopoulos MA, Avet-Loiseau H, et al. Prognostic value of minimal residual disease negativity in myeloma: combined analysis of POLLUX, CASTOR, ALCYONE, and MAIA. Blood. 2022;139:835–44.
Zamagni E, Nanni C, Dozza L, Carlier T, Bailly C, Tacchetti P, et al. Standardization of (18)F-FDG-PET/CT according to Deauville criteria for metabolic complete response definition in newly diagnosed multiple myeloma. J Clin Oncol. 2021;39:116–25.
Mateos MV, Kumar S, Dimopoulos MA, Gonzalez-Calle V, Kastritis E, Hajek R, et al. International Myeloma Working Group risk stratification model for smoldering multiple myeloma (SMM). Blood Cancer J. 2020;10:102.
Caers J, Paiva B, Zamagni E, Leleu X, Blade J, Kristinsson SY, et al. Diagnosis, treatment, and response assessment in solitary plasmacytoma: updated recommendations from a European Expert Panel. J Hematol Oncol. 2018;11:10.
Paiva B, Chandia M, Vidriales MB, Colado E, Caballero-Velazquez T, Escalante F, et al. Multiparameter flow cytometry for staging of solitary bone plasmacytoma: new criteria for risk of progression to myeloma. Blood. 2014;124:1300–3.
Basset M, Hummedah K, Kimmich C, Veelken K, Dittrich T, Brandelik S, et al. Localized immunoglobulin light chain amyloidosis: novel insights including prognostic factors for local progression. Am J Hematol. 2020;95:1158–69.
Sykes DB, O’Connell C, Schroyens W. The TEMPI syndrome. Blood. 2020;135:1199–203.
Afra TP, Namitha R, Khader A, Bishnoi A, Shahul Hameed DK, Razmi TM. Adenopathy and extensive skin patch overlying a plasmacytoma (AESOP) syndrome: erythrocyanotic vascular patches associated with an underlying rib plasmacytoma. Br J Dermatol. 2020;183:e33.
Kikkeri NN, Lazzi S, Santi R, Granai M, Onyango N, Leoncini L. A refined approach to the diagnosis of Burkitt lymphoma in a resource-poor setting. Histopathology. 2022;80:743–8.
Falini B, Martelli MP, Bolli N, Bonasso R, Ghia E, Pallotta MT, et al. Immunohistochemistry predicts nucleophosmin (NPM) mutations in acute myeloid leukemia. Blood. 2006;108:1999–2005.
Falini B, Mason DY. Proteins encoded by genes involved in chromosomal alterations in lymphoma and leukemia: clinical value of their detection by immunocytochemistry. Blood. 2002;99:409–26.
Acknowledgements
This work was supported by the Associazione Umbra contro le Leucemie e Linfomi (AULL).
Author information
Authors and Affiliations
Contributions
BF had the original idea and wrote the manuscript. GM and SL contributed to write the manuscript.
Corresponding author
Ethics declarations
Competing interests
BF is co-signer of the ICC classification of lymphoid neoplasms. SL is co-signer of the 5th WHO classification of lymphoid neoplasms. GM has no relevant conflict of interest concerning this report.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Falini, B., Martino, G. & Lazzi, S. A comparison of the International Consensus and 5th World Health Organization classifications of mature B-cell lymphomas. Leukemia 37, 18–34 (2023). https://doi.org/10.1038/s41375-022-01764-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-022-01764-1
