
Engineering Cardiac Small Extracellular Vesicle-Derived Vehicles with Thin-Film Hydration for Customized microRNA Loading

Abstract
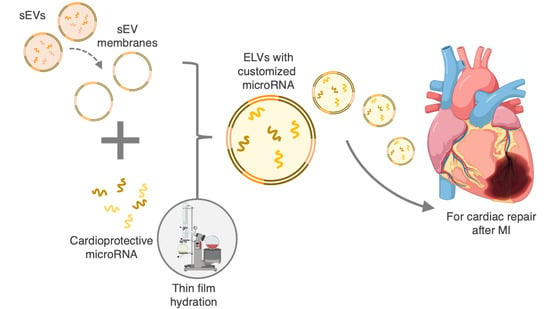
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Isolation and Culture of Human CPCs
2.2. Culture of Rat CECs
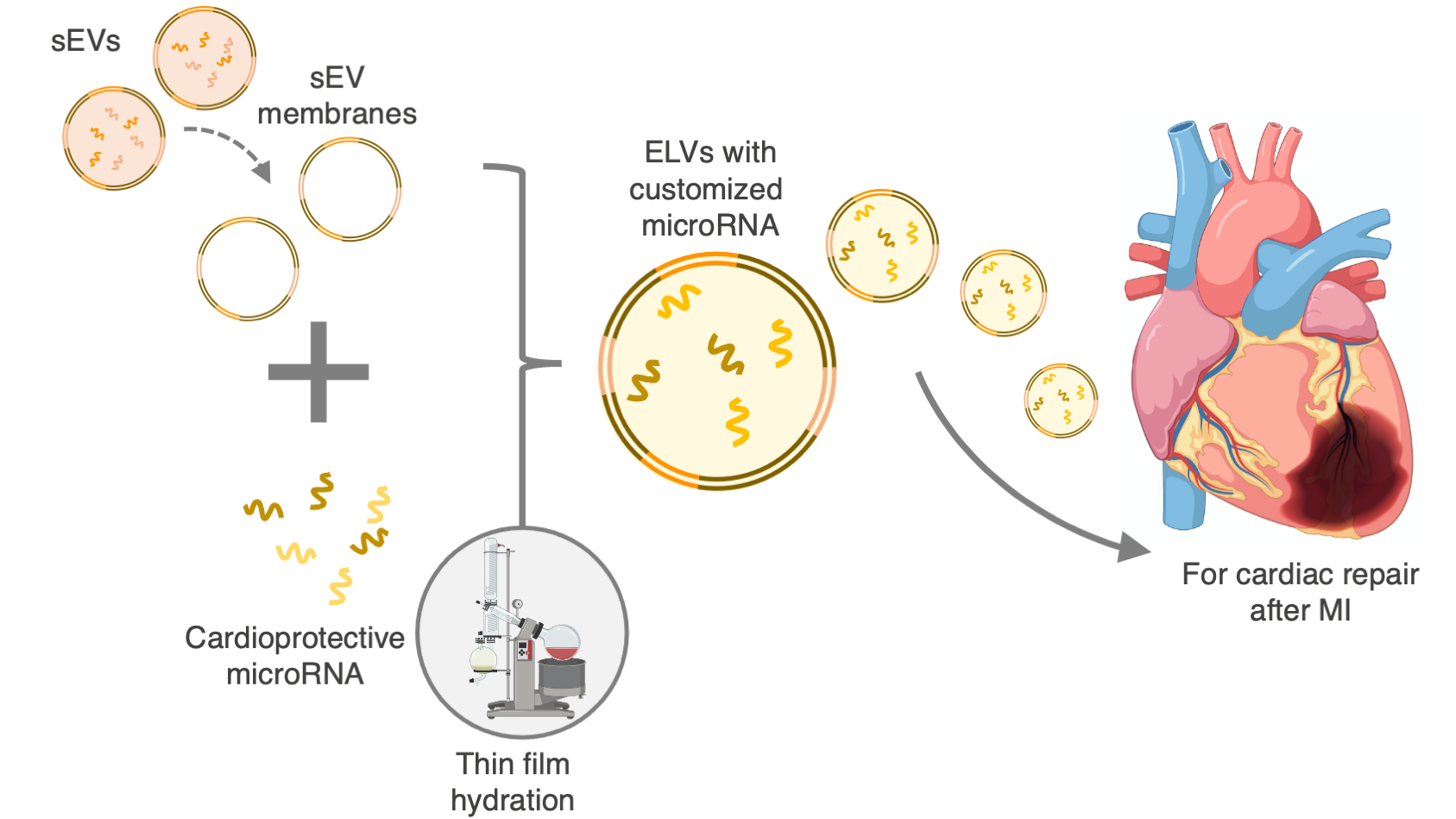
2.3. Isolation and Characterization of sEVs
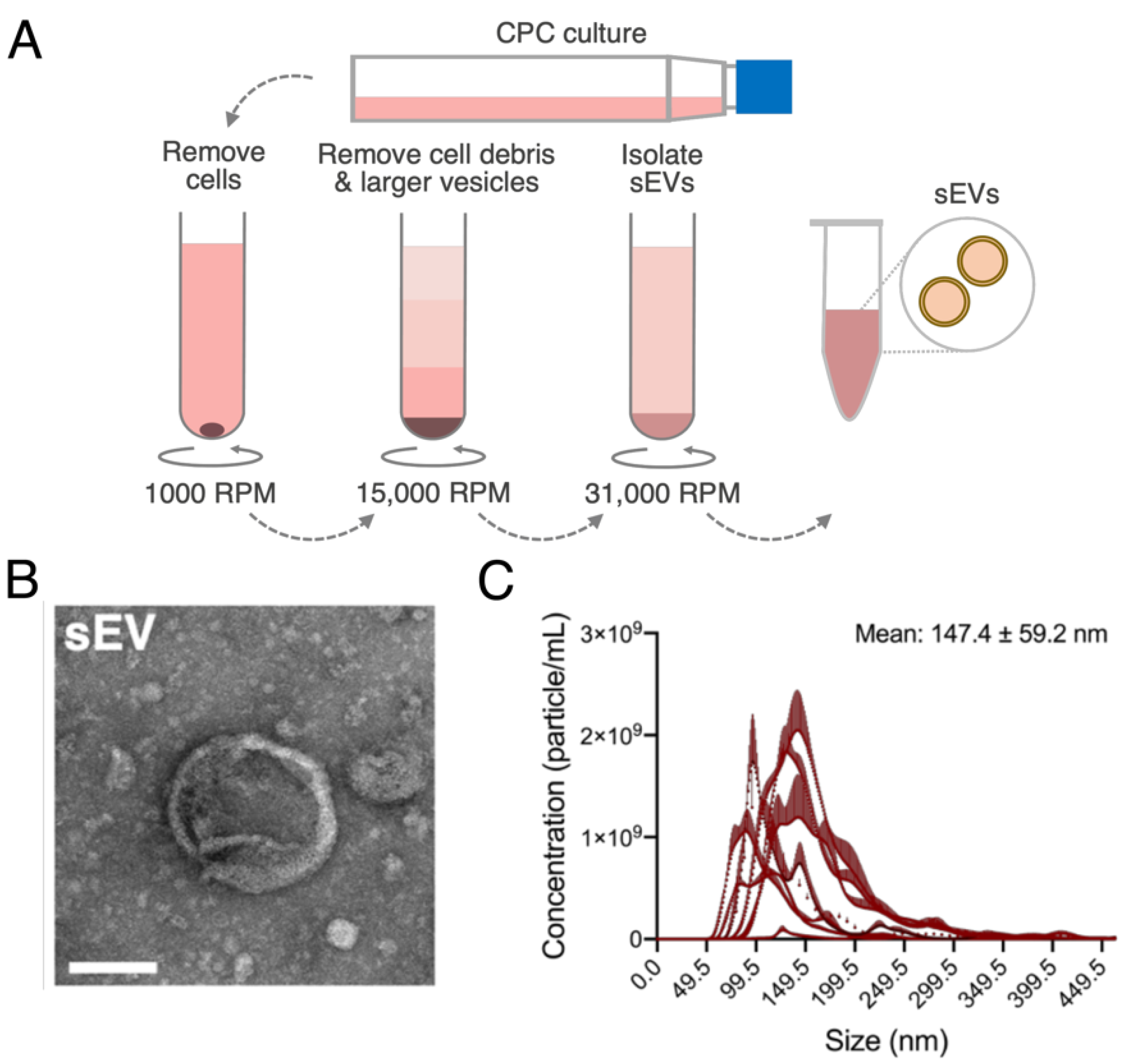
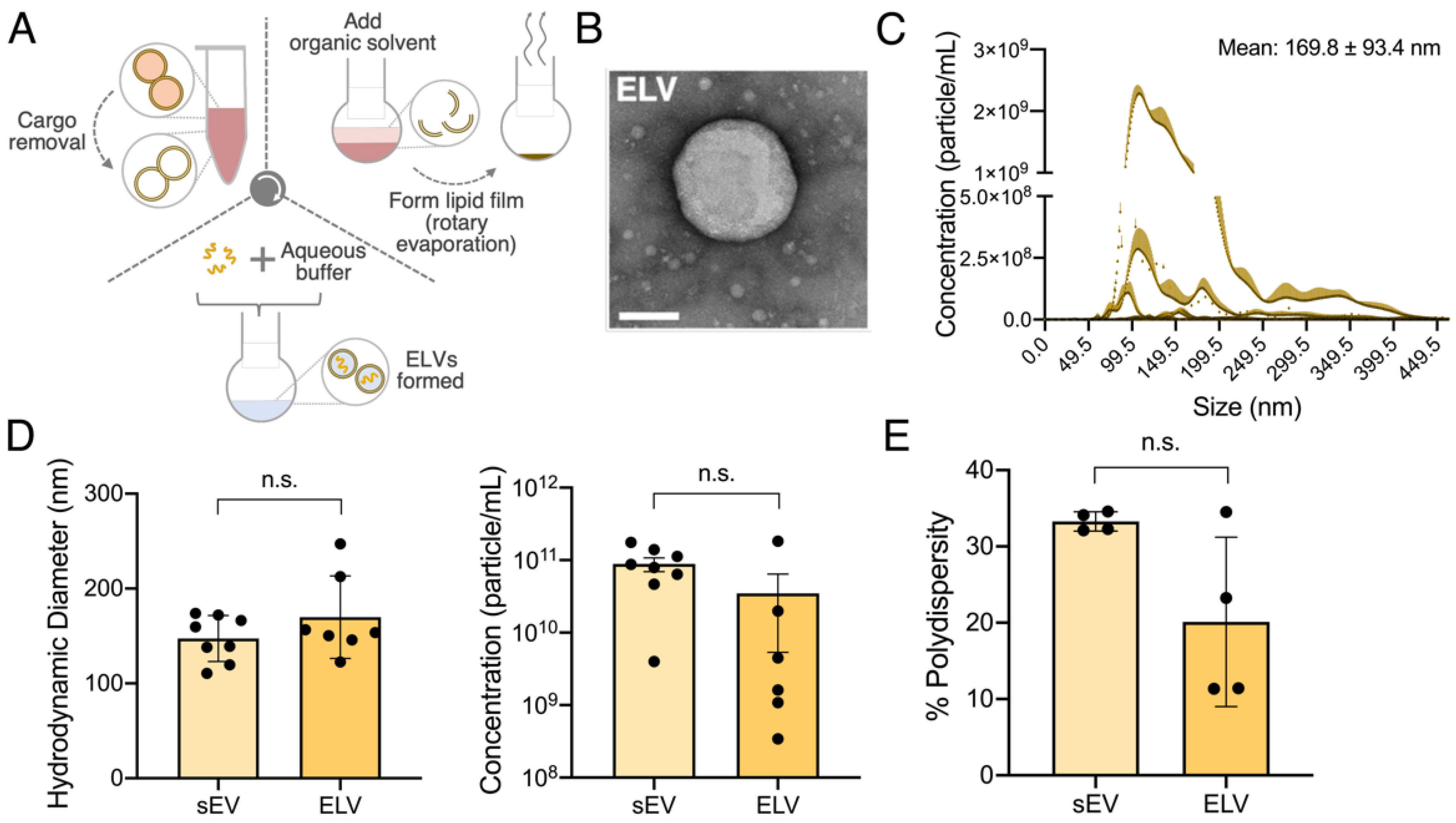
2.4. Formation of sEV Lipid Bilayer
2.5. Synthesis of miR-loaded ELVs
2.6. RNA Isolation and ELV Cargo Quantification
2.7. ELV Internalization
2.8. Tube Formation Assay
2.9. Statistical Analysis
3. Results
3.1. sEVs Successfully Isolated and Characterized from 2D CPC Cultures
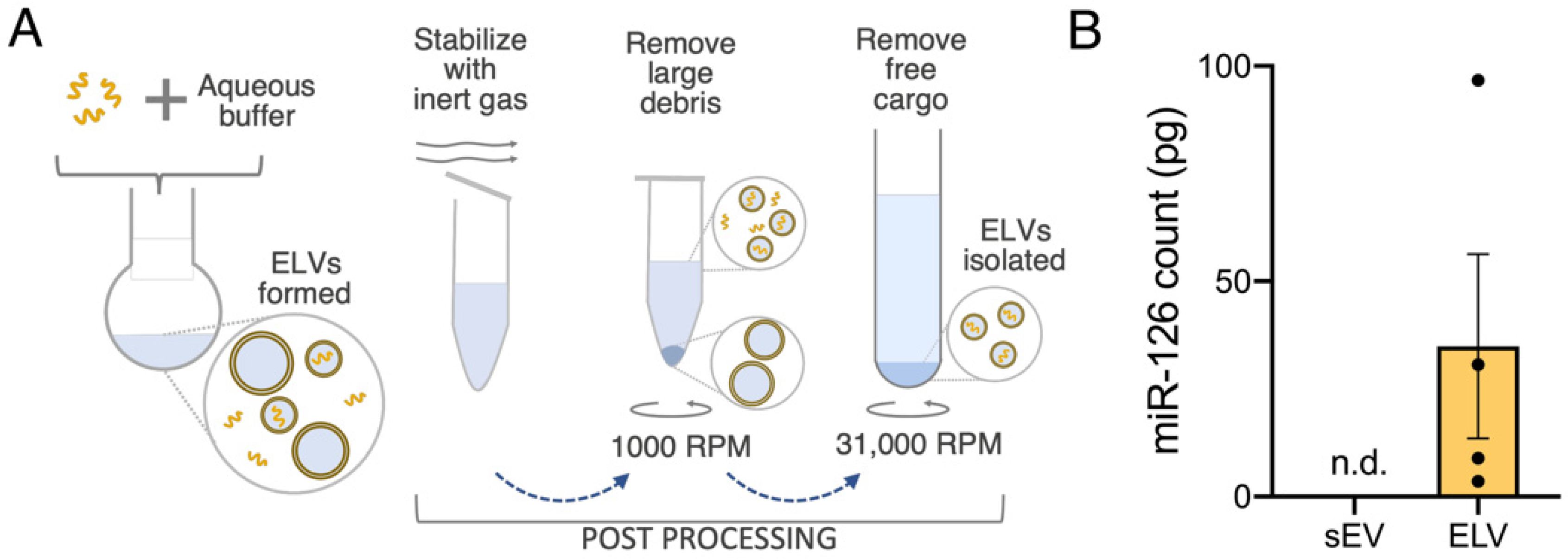
3.2. ELVs on A sEV Scale Successfully Synthesized with TFH with Selective miR Loading
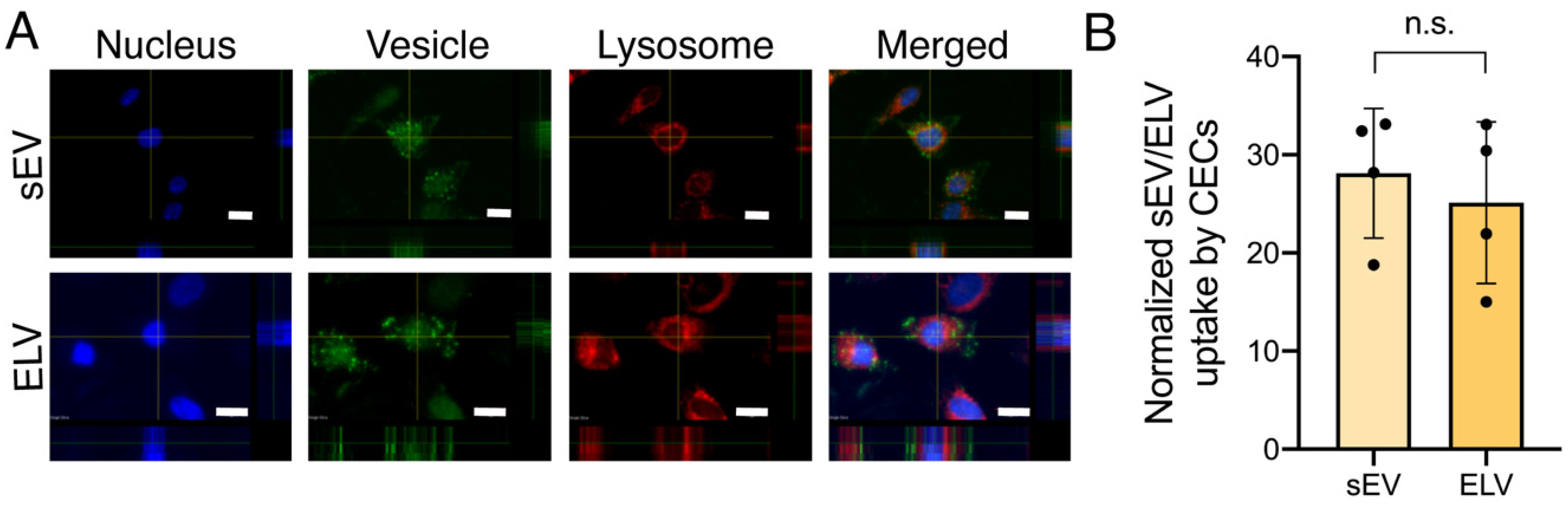
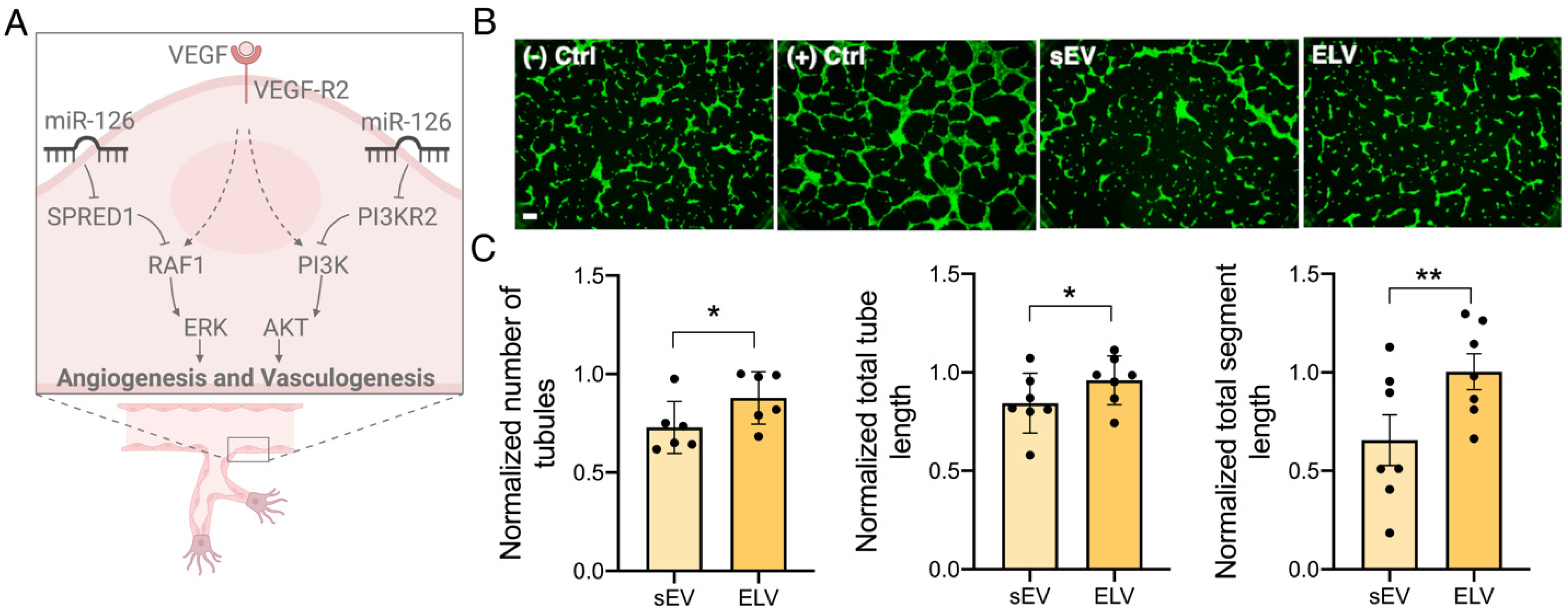
3.3. miR-126+ ELVs Are Taken up by CECs and Increase CEC Tube Formation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Aparicio, H.J.; Benjamin, E.J.; Callaway, C.W.; Carson, A.P.; Cheng, S.; Elkind, M.S.V.; Evenson, K.R.; Ferguson, J.F.; Knutson, K.L.; Lee, C.D.; et al. Heart Disease and Stroke Statistics-2021 Update: A Report from the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef]
- Bialik, S.; Geenen, D.L.; Sasson, I.E.; Cheng, R.; Horner, J.W.; Evans, S.M.; Lord, E.M.; Koch, C.J.; Kitsis, R.N. Myocyte apoptosis during acute myocardial infarction in the mouse localizes to hypoxic regions but occurs independently of p53. J. Clin. Investig. 1997, 100, 1363–1372. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Pathophysiology of myocardial infarction. Compr. Physiol. 2015, 5, 1841–1875. [Google Scholar] [PubMed]
- Li, Z.; Solomonidis, E.G.; Meloni, M.; Taylor, R.S.; Duffin, R.; Dobie, R.; Magalhaes, M.S.; Henderson, B.E.P.; Louwe, P.A.; D’Amico, G.; et al. Single-cell transcriptome analyses reveal novel targets modulating cardiac neovascularization by resident endothelial cells following myocardial infarction. Eur. Heart J. 2019, 40, 2507–2520. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Liu, M.; Sun, R.; Zheng, Y.; Zhang, P. Myocardial Infarction: Symptoms and Treatments. Cell Biochem. Biophys. 2015, 72, 865–867. [Google Scholar] [CrossRef] [PubMed]
- Shafei, A.E.S.; Ali, M.A.; Ghanem, H.G.; Shehata, A.I.; Abdelgawad, A.A.; Handal, H.R.; Talaat, K.A.; Ashaal, A.E.; El-Shal, A.S. Mesenchymal stem cell therapy: A promising cell-based therapy for treatment of myocardial infarction. J. Gene Med. 2017, 19, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Yeghiazarians, Y. Cardiac stem cell therapy: Review of the native cardiac progenitor cells and future direction. J. Cardiovasc. Pharmacol. 2014, 63, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Puddighinu, G.; D’Amario, D.; Foglio, E.; Manchi, M.; Siracusano, A.; Pontemezzo, E.; Cordella, M.; Facchiano, F.; Pellegrini, L.; Mangoni, A.; et al. Molecular mechanisms of cardioprotective effects mediated by transplanted cardiac ckit(+) cells through the activation of an inflammatory hypoxia-dependent reparative response. Oncotarget 2017, 9, 937–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dergilev, K.; Tsokolaeva, Z.; Makarevich, P.; Beloglazova, I.; Zubkova, E.; Boldyreva, M.; Ratner, E.; Dyikanov, D.; Menshikov, M.; Ovchinnikov, A.; et al. C-Kit Cardiac Progenitor Cell Based Cell Sheet Improves Vascularization and Attenuates Cardiac Remodeling following Myocardial Infarction in Rats. Biomed Res. Int. 2018, 2018, 3536854. [Google Scholar] [CrossRef]
- Bolli, R.; Hare, J.M.; March, K.L.; Pepine, C.J.; Willerson, J.T.; Perin, E.C.; Yang, P.C.; Henry, T.D.; Traverse, J.H.; Mitrani, R.D.; et al. Rationale and Design of the CONCERT-HF Trial (Combination of Mesenchymal and c-kit(+) Cardiac Stem Cells As Regenerative Therapy for Heart Failure). Circ. Res. 2018, 122, 1703–1715. [Google Scholar] [CrossRef]
- Barile, L.; Cervio, E.; Lionetti, V.; Milano, G.; Ciullo, A.; Biemmi, V.; Bolis, S.; Altomare, C.; Matteucci, M.; Di Silvestre, D.; et al. Cardioprotection by cardiac progenitor cell-secreted exosomes: Role of pregnancy-associated plasma protein-A. Cardiovasc. Res. 2018, 114, 992–1005. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.; Wang, L.; Li, Q.; Xu, J.; Xu, J.; Xiong, Y.; Chen, G.; Qian, H.; Jin, C.; Yu, Y.; et al. Combinatorial treatment of acute myocardial infarction using stem cells and their derived exosomes resulted in improved heart performance. Stem Cell Res. Ther. 2019, 10, 300. [Google Scholar] [CrossRef] [Green Version]
- Dykes, I.M. Exosomes in Cardiovascular Medicine. Cardiol. Ther. 2017, 6, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.G.E.; Cheng, K.; Marbán, E. Exosomes as critical agents of cardiac regeneration triggered by cell therapy. Stem Cell Rep. 2014, 2, 606–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, P.; Sharma, S.; Korutla, L.; Datla, S.R.; Shoja-Taheri, F.; Mishra, R.; Bigham, G.E.; Sarkar, M.; Morales, D.; Bittle, G.; et al. Circulating exosomes derived from transplanted progenitor cells aid the functional recovery of ischemic myocardium. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.; Klychko, E.; Thorne, T.; Misener, S.; Schultz, K.M.; Millay, M.; Ito, A.; Liu, T.; Kamide, C.; Agrawal, H.; et al. Exosomes from human CD34(+) stem cells mediate their proangiogenic paracrine activity. Circ. Res. 2011, 109, 724–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trac, D.; Hoffman, J.R.; Bheri, S.; Maxwell, J.T.; Platt, M.O.; Davis, M.E. Predicting Functional Responses of Progenitor Cell Exosome Potential with Computational Modeling. Stem Cells Transl. Med. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosme, J.; Guo, H.; Hadipour-Lakmehsari, S.; Emili, A.; Gramolini, A.O. Hypoxia-Induced Changes in the Fibroblast Secretome, Exosome, and Whole-Cell Proteome Using Cultured, Cardiac-Derived Cells Isolated from Neonatal Mice. J. Proteome Res. 2017, 16, 2836–2847. [Google Scholar] [CrossRef]
- Agarwal, U.; George, A.; Bhutani, S.; Ghosh-Choudhary, S.; Maxwell, J.T.; Brown, M.E.; Mehta, Y.; Platt, M.O.; Liang, Y.; Sahoo, S.; et al. Experimental, systems, and computational approaches to understanding the MicroRNA-mediated reparative potential of cardiac progenitor cell-derived exosomes from pediatric patients. Circ. Res. 2017, 120, 701–712. [Google Scholar] [CrossRef] [Green Version]
- Gray, W.D.; French, K.M.; Ghosh-Choudhary, S.; Maxwell, J.T.; Brown, M.E.; Platt, M.O.; Searles, C.D.; Davis, M.E. Identification of therapeutic covariant microRNA clusters in hypoxia-treated cardiac progenitor cell exosomes using systems biology. Circ. Res. 2015, 116, 255–263. [Google Scholar] [CrossRef] [Green Version]
- Chevillet, J.R.; Kang, Q.; Ruf, I.K.; Briggs, H.A.; Vojtech, L.N.; Hughes, S.M.; Cheng, H.H.; Arroyo, J.D.; Meredith, E.K.; Gallichotte, E.N.; et al. Quantitative and stoichiometric analysis of the microRNA content of exosomes. Proc. Natl. Acad. Sci. USA 2014, 111, 14888–14893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.S.; Haney, M.J.; Zhao, Y.; Mahajan, V.; Deygen, I.; Klyachko, N.L.; Inskoe, E.; Piroyan, A.; Sokolsky, M.; Okolie, O.; et al. Development of exosome-encapsulated paclitaxel to overcome MDR in cancer cells. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 655–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascucci, L.; Coccè, V.; Bonomi, A.; Ami, D.; Ceccarelli, P.; Ciusani, E.; Viganò, L.; Locatelli, A.; Sisto, F.; Doglia, S.M.; et al. Paclitaxel is incorporated by mesenchymal stromal cells and released in exosomes that inhibit in vitro tumor growth: A new approach for drug delivery. J. Control. Release 2014, 192, 262–270. [Google Scholar] [CrossRef]
- Wahlgren, J.; Karlson, T.D.L.; Brisslert, M.; Vaziri Sani, F.; Telemo, E.; Sunnerhagen, P.; Valadi, H. Plasma exosomes can deliver exogenous short interfering RNA to monocytes and lymphocytes. Nucleic Acids Res. 2012, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuhrmann, G.; Serio, A.; Mazo, M.; Nair, R.; Stevens, M.M. Active loading into extracellular vesicles significantly improves the cellular uptake and photodynamic effect of porphyrins. J. Control. Release 2015, 205, 35–44. [Google Scholar] [CrossRef]
- Haney, M.J.; Klyachko, N.L.; Zhao, Y.; Gupta, R.; Plotnikova, E.G.; He, Z.; Patel, T.; Piroyan, A.; Sokolsky, M.; Kabanov, A.V.; et al. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J. Control. Release 2015, 207, 18–30. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Böttger, R.; Qin, Z.; Kulkarni, J.A.; Vogler, J.; Cullis, P.R.; Li, S.-D. Phospholipid-Free Small Unilamellar Vesicles for Drug Targeting to Cells in the Liver. Small 2019, 15, e1901782. [Google Scholar] [CrossRef]
- Jung, H.T. The origins of stability of spontaneous vesicles. Proc. Natl. Acad. Sci. USA 2001, 98, 1353–1357. [Google Scholar] [CrossRef]
- Drulis-Kawa, Z.; Dorotkiewicz-Jach, A. Liposomes as delivery systems for antibiotics. Int. J. Pharm. 2010, 387, 187–198. [Google Scholar] [CrossRef]
- Ohno, S.I.; Drummen, G.P.C.; Kuroda, M. Focus on extracellular vesicles: Development of extracellular vesicle-based therapeutic systems. Int. J. Mol. Sci. 2016, 17, 172. [Google Scholar] [CrossRef] [Green Version]
- Grimaldi, N.; Andrade, F.; Segovia, N.; Ferrer-Tasies, L.; Sala, S.; Veciana, J.; Ventosa, N. Lipid-based nanovesicles for nanomedicine. Chem. Soc. Rev. 2016, 45, 6520–6545. [Google Scholar] [CrossRef] [Green Version]
- Bheri, S.; Hoffman, J.R.; Park, H.-J.; Davis, M.E. Biomimetic nanovesicle design for cardiac tissue repair. Nanomedicine 2020, 15, 1873–1896. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, K.B.; Gudbergsson, J.M.; Skov, M.N.; Christiansen, G.; Gurevich, L.; Moos, T.; Duroux, M. Evaluation of electroporation-induced adverse effects on adipose-derived stem cell exosomes. Cytotechnology 2016, 68, 2125–2138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Huang, H.; Zhang, L.; Bai, Y.; Chen, H. PCM and TAT co-modified liposome with improved myocardium delivery: In vitro and in vivo evaluations. Drug Deliv. 2017, 24, 339–345. [Google Scholar] [CrossRef] [Green Version]
- Song, T.; Wang, H.; Liu, Y.; Cai, R.; Yang, D.; Xiong, Y. TPGS-Modified Long-Circulating Liposomes Loading Ziyuglycoside I for Enhanced Therapy of Myelosuppression. Int. J. Nanomed. 2021, 16, 6281–6295. [Google Scholar] [CrossRef]
- Tamaddon, L.; Mohamadi, N.; Bavarsad, N. Preparation and Characterization of Mucoadhesive Loratadine Nanoliposomes for Intranasal Administration. Turk. J. Pharm. Sci. 2021, 18, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Abeesh, P.; Vishnu, W.K.; Guruvayoorappan, C. Preparation and characterization of withaferin A loaded pegylated nanoliposomal formulation with high loading efficacy: In vitro and in vivo anti-tumour study. Mater. Sci. Eng. C Mater. Biol. Appl. 2021, 128, 112335. [Google Scholar] [CrossRef]
- Agarwal, U.; Smith, A.W.; French, K.M.; Boopathy, A.V.; George, A.; Trac, D.; Brown, M.E.; Shen, M.; Jiang, R.; Fernandez, J.D.; et al. Age-Dependent Effect of Pediatric Cardiac Progenitor Cells After Juvenile Heart Failure. Stem Cells Transl. Med. 2016, 5, 883–892. [Google Scholar] [CrossRef]
- Cvjetkovic, A.; Lötvall, J.; Lässer, C. The influence of rotor type and centrifugation time on the yield and purity of extracellular vesicles. J. Extracell. Vesicles 2014, 3. [Google Scholar] [CrossRef]
- Rasband, W.S. ImageJ. U.S. National Institutes of Health. Available online: https://imagej.nih.gov/ij/ (accessed on 5 October 2021).
- Carpentier, G.; Berndt, S.; Ferratge, S.; Rasband, W.; Cuendet, M.; Uzan, G.; Albanese, P. Angiogenesis Analyzer for ImageJ—A comparative morphometric analysis of ‘Endothelial Tube Formation Assay’ and ‘Fibrin Bead Assay’. Sci. Rep. 2020, 10, 11568. [Google Scholar] [CrossRef]
- Wang, W.; Zheng, Y.; Wang, M.; Yan, M.; Jiang, J.; Li, Z. Exosomes derived miR-126 attenuates oxidative stress and apoptosis from ischemia and reperfusion injury by targeting ERRFI1. Gene 2019, 690, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Evers, M.J.W.; van de Wakker, S.I.; de Groot, E.M.; de Jong, O.G.; Gitz-François, J.J.J.; Seinen, C.S.; Sluijter, J.P.G.; Schiffelers, R.M.; Vader, P. Functional siRNA Delivery by Extracellular Vesicle–Liposome Hybrid Nanoparticles. Adv. Healthc. Mater. 2021, 2101202. [Google Scholar] [CrossRef] [PubMed]
- Corbeil, D.; Santos, M.F.; Karbanová, J.; Kurth, T.; Rappa, G.; Lorico, A. Uptake and Fate of Extracellular Membrane Vesicles: Nucleoplasmic Reticulum-Associated Late Endosomes as a New Gate to Intercellular Communication. Cells 2020, 9, 1931. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, A.; Takahashi, Y.; Nishikawa, M.; Sano, K.; Morishita, M.; Charoenviriyakul, C.; Saji, H.; Takakura, Y. Role of Phosphatidylserine-Derived Negative Surface Charges in the Recognition and Uptake of Intravenously Injected B16BL6-Derived Exosomes by Macrophages. J. Pharm. Sci. 2017, 106, 168–175. [Google Scholar] [CrossRef] [Green Version]
- Parikh, M.; Pierce, G.N. A Brief Review on the Biology and Effects of Cellular and Circulating microRNAs on Cardiac Remodeling after Infarction. Int. J. Mol. Sci. 2021, 22, 4995. [Google Scholar] [CrossRef]
- Moghaddam, A.S.; Afshari, J.T.; Esmaeili, S.A.; Saburi, E.; Joneidi, Z.; Momtazi-Borojeni, A.A. Cardioprotective microRNAs: Lessons from stem cell-derived exosomal microRNAs to treat cardiovascular disease. Atherosclerosis 2019, 285, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Pan, Q.; Wang, Y.; Lan, Q.; Wu, W.; Li, Z.; Ma, X.; Yu, L. Exosomes Derived from Mesenchymal Stem Cells Ameliorate Hypoxia/Reoxygenation-Injured ECs via Transferring MicroRNA-126. Stem Cells Int. 2019, 2019, 2831756. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Tian, X.; Ruan, Y.; Xing, J.; Meng, Z. Asiatic acid alleviates Ang-II induced cardiac hypertrophy and fibrosis via miR-126/PIK3R2 signaling. Nutr. Metab. 2021, 18, 71. [Google Scholar] [CrossRef]
- Yang, H.-H.; Chen, Y.; Gao, C.-Y.; Cui, Z.-T.; Yao, J.-M. Protective Effects of MicroRNA-126 on Human Cardiac Microvascular Endothelial Cells Against Hypoxia/Reoxygenation-Induced Injury and Inflammatory Response by Activating PI3K/Akt/eNOS Signaling Pathway. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 42, 506–518. [Google Scholar] [CrossRef]
- Harris, T.A.; Yamakuchi, M.; Ferlito, M.; Mendell, J.T.; Lowenstein, C.J. MicroRNA-126 regulates endothelial expression of vascular cell adhesion molecule 1. Proc. Natl. Acad. Sci. USA 2008, 105, 1516–1521. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bheri, S.; Kassouf, B.P.; Park, H.-J.; Hoffman, J.R.; Davis, M.E. Engineering Cardiac Small Extracellular Vesicle-Derived Vehicles with Thin-Film Hydration for Customized microRNA Loading. J. Cardiovasc. Dev. Dis. 2021, 8, 135. https://doi.org/10.3390/jcdd8110135
Bheri S, Kassouf BP, Park H-J, Hoffman JR, Davis ME. Engineering Cardiac Small Extracellular Vesicle-Derived Vehicles with Thin-Film Hydration for Customized microRNA Loading. Journal of Cardiovascular Development and Disease. 2021; 8(11):135. https://doi.org/10.3390/jcdd8110135
Chicago/Turabian StyleBheri, Sruti, Brandon P. Kassouf, Hyun-Ji Park, Jessica R. Hoffman, and Michael E. Davis. 2021. "Engineering Cardiac Small Extracellular Vesicle-Derived Vehicles with Thin-Film Hydration for Customized microRNA Loading" Journal of Cardiovascular Development and Disease 8, no. 11: 135. https://doi.org/10.3390/jcdd8110135