
Role of Oxidative Stress in Ocular Diseases: A Balancing Act
by
 , , , , , and
, , , , , and
Daisy Y. Shu
1 ,
,
Suman Chaudhary
1,
Kin-Sang Cho
1,
Anton Lennikov
1,
William P. Miller
1,
David C. Thorn
2,
Menglu Yang
1 and
Tina B. McKay
3,* 1
Department of Ophthalmology, Schepens Eye Research Institute of Mass Eye and Ear, Harvard Medical School, Boston, MA 02114, USA
2
Department of Chemistry and Chemical Biology, Harvard University, Cambridge, MA 02138, USA
3
Department of Anesthesia, Critical Care and Pain Medicine, Massachusetts General Hospital, Harvard Medical School, Boston, MA 02114, USA
*
Author to whom correspondence should be addressed.
Metabolites 2023, 13(2), 187; https://doi.org/10.3390/metabo13020187
Submission received: 31 December 2022
/
Revised: 22 January 2023
/
Accepted: 24 January 2023
/
Published: 27 January 2023
(This article belongs to the Special Issue Bioenergetics and Cellular Dysfunction in the Brain and Eye)
Abstract
:Redox homeostasis is a delicate balancing act of maintaining appropriate levels of antioxidant defense mechanisms and reactive oxidizing oxygen and nitrogen species. Any disruption of this balance leads to oxidative stress, which is a key pathogenic factor in several ocular diseases. In this review, we present the current evidence for oxidative stress and mitochondrial dysfunction in conditions affecting both the anterior segment (e.g., dry eye disease, keratoconus, cataract) and posterior segment (age-related macular degeneration, proliferative vitreoretinopathy, diabetic retinopathy, glaucoma) of the human eye. We posit that further development of therapeutic interventions to promote pro-regenerative responses and maintenance of the redox balance may delay or prevent the progression of these major ocular pathologies. Continued efforts in this field will not only yield a better understanding of the molecular mechanisms underlying the pathogenesis of ocular diseases but also enable the identification of novel druggable redox targets and antioxidant therapies.
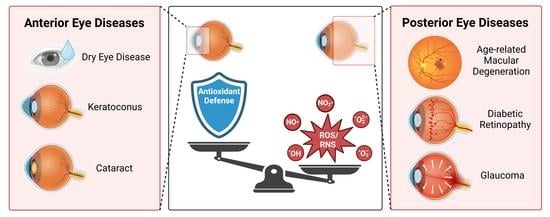
1. Introduction
Aging is a major risk factor for many diseases, including cancer, cardiovascular disease, and neurodegeneration [1,2]. As we age, our increased susceptibility to disease may partially be attributed to genetic and epigenetic changes that develop over time with continual environmental exposure and damage from endogenous and exogenous reactive oxygen species (ROS). Inept pro-reparative responses and telomere shortening are central to the underlying mechanisms involved in aging [3]. It is thought that the biological age of an organism is largely dependent on the body’s ability to manage internal and external stress at the cellular level, which may lead to cell dysfunction if left uncontrolled. While the gradual deterioration in genomic stability and cellular senescence are inevitable with aging, mutations in mitochondrial DNA (mtDNA), altered proteostasis, and nutrient-sensing also contribute to disrupted bioenergetics, which influences tissue and organismal survival [4].
The human eye is not resistant to processes associated with aging. At the front of the eye, the cornea and lens are particularly susceptible to oxidative stress that can be attributed to direct exposure to ultraviolet (UV) light emitted by the sun. As a highly metabolic tissue, the retina is also at increased risk of age-associated processes that contribute to increased oxidative stress and neurodegeneration. Damage to these cell layers may cause deficits in visual acuity or progressive vision loss and significantly affect mobility and quality of life.
In this review, we introduce each pathological condition affecting the major tissues within the human eye and highlight the role of oxidative stress in their underlying disease etiologies. Understanding the conserved mechanisms involved in the disruption of the cellular redox balance may aid in developing targeted interventions to prevent or delay the progression of the major ocular pathologies associated with oxidative stress.
2. Sources of Oxidative Stress
A homeostatic balance exists between the production and quenching of ROS produced during normal physiological processes (Figure 1). The intracellular compartments of a cell maintain a reducing environment with a high abundance of glutathione (GSH) to prevent oxidative damage. The maintenance of this redox balance is essential in permitting downstream signaling processes that rely upon an oxidative state to activate secondary messengers. Of the sources of ROS, oxidative phosphorylation within the electron transport chain (ETC) in mitochondria is the major contributor to superoxide production, which may be converted to hydrogen peroxide via the catalytic activity of superoxide dismutase (SOD). Mitochondria perform essential functions within a cell, serving as primary energy generators in the form of ATP. Somatic mutations in mtDNA may develop gradually over time with age, leading to decreased mitochondrial function and energy output [5]. Mitochondria are thought to acquire mutations at a rate of 10–100X more often than mutations in the nuclear genome [6] due to the increased oxidative environment within the mitochondrion, the absence of protective histones for mtDNA, and the minimal proofreading capabilities of DNA polymerase γ that contribute to lower replication fidelity [7,8].
The metabolism of oxygen produces a low level of ROS in a beneficial, physiological process. Under normal conditions, the ETC promotes ATP synthesis by maintaining a voltage gradient across the inner mitochondrial membrane (ΔΨm) that drives ATP-synthase. Oxygen plays a critical role in this process by acting as the final electron acceptor, serving as a nucleophile for two protons to generate water. However, when ΔΨm is in excess, oxygen can also be partially reduced to form superoxide (O2·−), which can then be protonated to form other reactive species, including hydrogen peroxide (H2O2) and the hydroxyl radical (·OH). The oxidative phosphorylation pathway is executed by a series of protein complexes (complexes I–IV) embedded in the mitochondrial membrane [9]. Nicotinamide adenine dinucleotide (NADH2) and flavin adenine dinucleotide (FADH2) donate electrons that are transported between these protein complexes, coupled with the pumping of hydrogen ions across the mitochondrial membrane [9]. Electron leakage from complex I and complex III will react with oxygen and form ROS, including hydrogen peroxide, hydroxyl radicals, and other reactive oxygen and nitrogen species [10,11]. Other non-mitochondrial sources of free radicals are the nitric oxide synthase reaction and cytochrome P450 system, among others [12].
Excess intracellular ROS production can lead to the oxidation of DNA, proteins, lipids, and metabolites, thereby leading to possible disruptions in gene and protein expression patterns, protein aggregation, and cellular dysfunction. Endogenous ROS may also promote the generation of lipid hydroperoxides that function as key intermediates in ROS-mediated processes. The major biomarkers of oxidative stress reported in studies of the eye include malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE), which are byproducts of lipid peroxidation mediated via hydrogen peroxide, superoxide, or other ROS (Table 1). Cyclopentenone prostaglandins are also important mediators involved in lipid peroxidation that have been found to promote ROS production and cytotoxicity.
The cellular antioxidant defense system in most cell types consists of non-enzymatic and enzymatic components responsible for scavenging free radicals and non-radical oxidants [24]. Examples of intracellular non-enzymatic protective mechanisms against excess ROS are metal-binding proteins (albumin, ferritin, lactoferrin), retinol (Vitamin A), ascorbic acid (Vitamin C), tocopherol (Vitamin E), coenzyme Q10, and polyamines (spermine, spermidine, putrescine). The enzymatic protective mechanisms responsible for reacting with ROS include superoxide dismutase (SOD), catalase, peroxiredoxins, glutathione peroxidase, and glutathione reductase, among others. There are three main isoforms of SOD, characterized by their cellular localization: cytosolic (SOD1 (CuZn-SOD)), mitochondrial (SOD2 (Mn-SOD)), and extracellular (SOD3 (EC-CuZn-SOD)). Compared to other tissues, the eye has lower levels of SOD proteins, with the highest distribution present within the retina [25]. The transcription factor nuclear factor erythroid-related factor 2 (Nrf2) is another crucial element of the cellular antioxidant response [26,27]. Upon being activated by a change in the redox balance, it binds to enhancer regions in the promoters of antioxidant genes, known as antioxidant response elements, to facilitate their transcription and generate pools of antioxidant proteins to combat cellular ROS.
Arachidonic acid (ARA) and its metabolites play a crucial role in inflammation and the regulation of oxidative stress. While oxidative stress can enhance ARA mobilization [28], ARA itself can potentiate or, in some cases, confer resistance to oxidative stress. This 20-carbon chain fatty acid belongs to the group of omega-6 (n-6) polyunsaturated fatty acids (PUFAs). In its esterified form, ARA is found covalently bound to membrane phospholipids, enabling the fluidity and flexibility of cell membranes. The stimulation of specific cell-surface receptors activates phospholipase A2, triggering the release of the free form of ARA from the cell membrane during inflammation. ARA is then metabolized by enzymes such as cyclooxygenases, lipoxygenases, and cytochrome P450 to generate a spectrum of inflammatory bioactive mediators, including prostaglandins, leukotrienes, and lipoxins [29].
While the role of ARA and oxidative stress has not been well studied in ocular tissues, we can glean evidence of this relationship from other fields. During lipoperoxidation, fatty acids within membrane phospholipids undergo oxidative modification, altering membrane fluidity and resulting in dysregulated cellular signaling and aberrant protein structures [28]. The repair of these phospholipids requires the selective cleavage of peroxidized fatty acid residues and their replacement by native fatty acids. This process can enhance phospholipase A2 activity, which facilitates ARA mobilization, leading to enhanced inflammatory signaling [30]. In adult cardiac fibroblasts, hydrogen peroxide was found to activate Nox4 through phospholipase A2-dependent ARA production [31]. While oxidative stress can mobilize ARA through Ca2+-dependent and Ca2+-independent phospholipase A2 mechanisms, free ARA can also be generated by alternative, phospholipase A2-independent pathways, such as through the inhibition of fatty acid reacylation. For example, the exposure of rat alveolar macrophages [32] and vascular smooth muscle cells [33] to hydrogen peroxide resulted in the impairment of fatty acid esterification and the subsequent accumulation of free ARA.
Conversely, ARA itself can activate key oxidative stress molecules. In HeLa cells, ARA was found to induce the interaction of the p67phox-Rac complex with Nox2, leading to the production of superoxide [34]. Intriguingly, in hippocampal slices, ARA has been shown to exhibit neuroprotective effects by defending against oxidative stress through the enhancement of the internal antioxidant system [35]. Further research is required to explore the precise relationship between ARA and oxidative stress in ocular diseases.
3. Cornea
The cornea is a transparent avascular tissue layer of about 500 μm thickness located at the front of the eye [36]. The cornea is composed of three primary cellular layers: a stratified epithelium, stromal keratocytes, and a single-layered endothelium. The corneal epithelium forms an external cellular surface, which acts as the frontline defense against environmental pathogens, chemical irritants, and abrasive injury. Corneal epithelial cells are exposed to high concentrations of oxygen and UV radiation and thus require powerful antioxidant defenses to prevent oxidative damage. Within the corneal stroma, corneal keratocytes reside in, secrete, and assemble the surrounding extracellular matrix. The bulk mass of the cornea is attributed to the extracellular matrix of the stroma, primarily composed of collagen types I and V and the proteoglycans keratocan, decorin, lumican, and mimecan, which are important in limiting the collagen fibril diameter and maintaining tissue transparency [37]. There are also a number of resident immune cells that have been detected within the uninjured cornea, including macrophages, dendritic cells, and a small population of γδ T cells [38]. Here, we discuss the role of elevated oxidative stress in two of the most common pathologies affecting the cornea, dry eye disease and keratoconus (KC).
3.1. Dry Eye Disease
Dry eye is a multifactorial disease characterized by ocular dryness, discomfort, pain, and tear film instability. Current evidence suggests that ROS generated from environmental factors, such as UV radiation and pollution, contribute to the pathogenesis of dry eye disease [39,40]. These findings correlate with a strong association between dry eye and aging, as excess ROS accumulates with older age [41]. Molecular markers of oxidative stress, such as 8-hydroxy-2-deoxyguanosine (8-OHdG), 4-HNE, and MDA, have been observed in a blink-suppressed dry eye model [42]. 4-HNE and another biomarker of oxidative stress, hexanoyl-lysine, was found to be significantly higher in the conjunctiva of patients with Sjögren’s syndrome and dry eye compared to healthy controls [43]. Moreover, in mice with compromised antioxidant capacity induced by the knockout of SOD-1, atrophy of the lacrimal gland occurs, leading to decreased tear production and eventually dry eye [44]. This evidence supports an association between oxidative stress and dry eye disease in animal models and human clinical populations.
The tear film is a thin layer of liquid covering the corneal surface and conjunctiva, which comprises a lipid layer, an aqueous layer, and a mucous layer. The lipid layer is secreted by the meibomian gland located in the eyelids; the lacrimal gland contributes the bulk of the aqueous layer, and the mucous layer is made up of mucin secreted by conjunctival goblet cells and corneal epithelial cells. Disturbances to any of the three layers will lead to dry eye disease. The current literature on ROS in dry eye disease focuses primarily on the dysfunction of the lacrimal and meibomian glands. Uchino et al. observed lacrimal gland damage and decreased tear production in a tetracyclic-mev-1 conditional transgenic mouse model [45]. In this model, the mev-1 mutant has a genetic dysfunction in complex II of the mitochondrial ETC, leading to the overproduction of prooxidants by the mitochondria. Compared to the wild type, mice carrying the mutation showed increased mRNA expression of inflammatory markers, including tumor necrosis factor (TNF-α), interleukin (IL)-6, IL-1β, and interferon (IFN)-γ, as well as higher infiltration of CD4+ T cells, CD8+ T cells, B cells, and macrophages. Proinflammatory cytokines such as TNF-α have the capacity to produce ROS [46], further potentiating oxidative stress and perpetuating the vicious cycle of ROS accumulation. Damage to the normal structure of the acini was also present in the mutant mice, indicating the role of oxidative stress in causing deficient tear production by directly damaging the lacrimal gland [45].
In addition to direct structural damage, oxidative stress may disrupt the neural reflex arc of tear secretion. The cornea and conjunctiva are innervated by sensory nerve endings originating from the trigeminal ganglia. When the sensory nerve is stimulated, it conducts the signal to the central nervous system, activating the efferent parasympathetic and sympathetic nerves that innervate the acini of the lacrimal gland [47]. Prooxidants on the ocular surface damage the myelin of the afferent sensory nerve, causing reduced signal transmission to the lacrimal gland, which in turn leads to decreased tear secretion [40].
The meibomian glands are sebaceous glands located inside the tarsal plates of both the upper and lower eyelids, with openings at the rims of the eyelids. They secrete meibum, which forms the lipid layer of the tear film. The instability of the lipid layer is known as meibomian gland dysfunction, which is a common cause of evaporative dry eye disease [48]. Meibomian gland dysfunction was observed in SOD1 knockout mice, whereby large lipid droplets were found inside meibomian glands, along with increased inflammatory markers (IL-6 and TNF-α) and the infiltration of CD45+ leukocytes [49]. These data indicate that a deficiency in antioxidant capacity may induce inflammation and structural damage to the meibomian gland.
Over the years, clinicians have attempted to treat dry eye disease with antioxidative agents, including Vitamin B12, iodide iontophoresis, and PUFAs. Patients using hyaluronic acid eye drops supplemented with Vitamin B12 reported significantly reduced levels of prooxidant markers in the tear film with longer tear film breakup times and lower Ocular Surface Disease Index scores [50]. It is noteworthy to mention that this study did not include a hyaluronic acid-only group, and thus, the beneficial effects of additional Vitamin B12 are debatable. Iodide is a reducing agent that can neutralize oxidants. Dry eye patients treated with iodide iontophoresis demonstrated significant improvement in dry eye symptoms and clinical evaluation compared to the control group, where participants were given iodide without an electrical current [51]. More recent studies have focused on the use of PUFAs, mainly omega-3 FAs, such as docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA). PUFAs are fatty acids in which the hydrocarbon chain contains two or more double bonds. These double bonds enable PUFAs to better neutralize ROS. However, according to a recent review of 34 randomized controlled trials involving more than 4314 adult participants, omega-3 FA intervention showed little to no beneficial effect on the symptomatic control of dry eye disease compared to placebo [52]. Therefore, further research is required to optimize the efficacy and route of administration of antioxidants in treating dry eye disease.
A recent result showed that melatonin, a hormone secreted by the pineal gland, protects corneal epithelial cells in a dry eye disease model [53]. The role of different hormone levels in the development of dry eye has been extensively discussed, including sex hormones [54,55], oxytocin [56], growth hormones [57], and many more. These recent studies provide new ideas for future research to investigate the role of different hormones in the maintenance of the ocular surface.
Moreover, further mechanistic research is necessary to determine the source of prooxidants observed in dry eye disease, as well as how dry eye disease may be induced by uncontrolled oxidative stress.
3.2. Altered Metabolism and Bioenergetics in Keratoconus
KC is a corneal ectasia that affects about 1 per 500 to 2000 people worldwide, depending on the geographical region [58]. KC is characterized by a thinning of the corneal stroma that leads to the protrusion of the central cornea, the development of severe astigmatism, and visual deficits. In terms of pathophysiology, growing evidence suggests that oxidative stress may play an important role in the underlying etiology of KC [59,60,61,62]. Elevated lipid peroxidation, levels of MDA, reactive oxygen and nitrogen species, and lower total antioxidant capacity have all been associated with KC [61]. Markers of oxidative stress have been detected in the tears [63,64], sera [65], stromal fibroblasts [66], and corneal tissue [67,68,69] of KC patients. KC is often diagnosed in adolescence and early adulthood and generally stabilizes in midlife [70]. Hence, elevated oxidative stress in the cornea is likely not a result of aging or cumulative DNA damage [71] but rather thought to be partly attributed to acquired defects in mitochondrial function or ROS-scavenging abilities, leading to broad cellular stress in the corneal stroma, decreased collagen expression, keratocyte loss, and, ultimately, corneal thinning [72]. Mutations in mitochondrial-associated genes and reduced levels of SOD1 have been associated with familial and sporadic KC [73,74,75,76,77,78], though SOD1 mutations do not appear to be a universal biomarker of KC in all clinical populations [79,80,81,82]. As recently reviewed, other gene variants associated with KC include lysyl oxidase, collagen types IV and V, and inner mitochondrial membrane peptidase [83].
Elevated proinflammatory factors, including IL-6, TNF-α, and matrix metalloproteinase (MMP)-9, have been detected in KC tears, with higher levels correlating with increased KC severity [84]. Untargeted mass spectrometry analysis of human serum from a small case–control study revealed increased dehydroepiandrosterone sulfate and prostaglandins in patients with KC compared to age-matched controls [85], which is consistent with a study by our group [86] and others [87,88] associating altered hormone levels or their receptors with KC. Growing evidence supports the hypothesis that there is a systemic component involved in KC pathogenesis upstream of the endogenous oxidative stress observed in the cornea [89,90].
In terms of functional differences, KC-derived corneal fibroblasts deposit a thinner extracellular matrix with higher profibrotic collagen type III deposition and lower collagen type I compared to controls [91,92]. Stem cells derived from the corneoscleral rim likewise display decreased proliferative ability and elevated α-smooth muscle actin (α-SMA) expression in KC-derived spheroids [93]. Collectively, these in vitro findings have been consistent with the phenotype of moderate to severe KC in vivo [94], suggesting that the genetic or epigenetic features of KC are retained in early passages of primary corneal stromal cells.
A recurring theme of the underlying KC pathophysiology is the presence of altered cellular metabolism and bioenergetics. A metabolomic study of isolated stromal tissue from patients with KC revealed moderate differences in metabolite levels associated with fatty acid metabolism, the tricarboxylic acid (TCA) cycle, and arginine and proline metabolism [95]. Metabolic differences have also been identified in KC-derived corneal fibroblasts, including differential cytosolic levels of metabolites important in glucose metabolism, lower arginine levels, and increased lactate production compared to controls [66]. We have found that treatment with the antioxidant quercetin modulates steady-state levels of metabolites involved in glycolysis and the TCA cycle and reduces lactate production in corneal fibroblasts [96]. Quercetin treatment also appears to possess antifibrotic properties characterized by decreased collagen type III and α-SMA expression in vitro [97] and the blunting of corneal scarring in vivo [98].
Riboflavin-mediated crosslinking is an FDA-approved treatment for KC that also promotes lower lactate/malate levels in tears [99] and reduced lactate production by corneal fibroblasts [100], providing a degree of stabilization to the metabolic phenotype observed in KC, in addition to stiffening the collagen matrix to blunt progressive thinning. Moreover, basal arginase activity and hydroxyproline levels have been found to be lower in KC-derived keratocytes [101], correlating with a reduction in collagen deposition associated with these cells [102]. Our group has found that arginine supplementation promotes a modest increase in collagen secretion by KC-derived corneal fibroblasts [103], suggesting that exogenous arginine may modulate certain deficiencies in procollagen building blocks and stimulate matrix production at the cellular level. Further investigations of antioxidants and their role in influencing tissue regeneration via metabolic regulation are needed to determine whether reducing excess ROS in the stroma or stimulating the perturbation of cellular metabolism during wound healing may mitigate fibrotic processes to favor wound healing and scarless matrix deposition in KC.
Shared pathological mechanisms have been proposed for KC and Fuchs’ endothelial corneal dystrophy (FECD) involving elevated intracellular oxidative stress and reduced antioxidant responses, as previously reviewed [104,105]. FECD primarily affects older individuals and is associated with oxidative stress within the corneal endothelium that is thought to lead to the formation of aggregated extracellular matrix components in the form of guttae, the loss of corneal endothelial cells, and severe corneal edema. Increased lipid peroxidation has been detected in corneal tissue from patients with FECD showing ROS-mediated damage and progressive apoptosis of corneal endothelial cells [106]. These findings suggest that excess oxidative stress due to reduced antioxidant capacity or increased mitochondrial dysfunction within the corneal stroma and endothelial layer in the case of KC or FECD, respectively, may contribute to the loss of corneal transparency and significant defects in visual acuity.
4. Oxidative Stress in Cataract
The ocular lens is a transparent biconvex tissue suspended behind the iris by the lens zonules. Its highly organized architecture comprises two cell types: an anterior monolayer of cuboidal lens epithelial cells (LECs) and a mass of elongated lens fiber cells (LFCs), all encapsulated within a thickened basement membrane, known as the lens capsule [107]. The loss of lens transparency is known as cataract, which is the leading cause of blindness worldwide [108]. There are various types of cataract based on their anatomical location (nuclear, cortical, and subcapsular) and/or their etiology (age-related, diabetic, radiation, traumatic, and nutritional), the most common being age-related nuclear (ARN) cataract [109]. Currently, the only treatment for cataract is through surgical intervention, and while this initially results in a good visual prognosis, 35% of patients develop posterior capsular opacification (PCO), also known as secondary cataract, requiring further intervention to restore vision [110].
The proper functioning and longevity of the eye lens is achieved via several trade-offs. The removal of subcellular organelles, including ribosomes, from LFCs enables transparency [111] at the cost of protein turnover [112]. Encapsulation and devascularization of the lens help protect it from oxidative stress—oxygen’s partial pressure in the lens core is extraordinarily low [113]—though at the expense of potentially more efficient delivery of cellular reductants [114]. Similarly, an abundance of crystallin proteins (α, β, and γ in vertebrates) confers the lens with a high refractive index [115]; however, crystallins must maintain their folded and soluble state throughout life to ensure lens transparency [116]. As mature and metabolically quiescent LFCs lose their ability to synthesize and replace old proteins, crystallins accumulate post-translational modifications with age. Among these biochemical modifications are oxidation (including cysteine thiolation and disulfide formation), truncation, glycation, deamidation, and aspartate isomerization [117], as well as non-disulfide covalent crosslinking [118]. Individually or cumulatively, age-related modifications destabilize crystallins, leading to their partial unfolding and conversion to large protein aggregates that scatter light and cause the lens opacification characteristic of ARN cataract [117]. This age-related process is accelerated by environmental stresses such as exposure to ultraviolet radiation [119] and heavy metals [120]; the UV-light-induced oxidation of tryptophan residues in crystallins is highly correlative, with levels of intrinsic tryptophan fluorescence providing a measurable and reliable indicator of cataract severity [121].
The oxidation of methionine and cysteine residues is the chief modification in crystallins associated with ARN cataract [122,123]. The extensive disulfide linkages in crystallin aggregates from late-stage cataractous lenses have been recognized for decades [124,125,126], although other modifications such as deamidation are also prevalent [127,128]. There is growing evidence that oxidative and non-oxidative modifications, including deamidation [129,130], as well as amino acid mutations associated with inheritable, early-onset forms of cataract [131], act synergistically to predispose crystallins to aggregation and cataract formation. The molecular mechanisms underlying the oxidative misfolding and aggregation of crystallins have not been elucidated in detail. However, it is evident from the three-dimensional structures of the homologous βγ-crystallins that disulfide formation between pairs of natively buried, spatially distant cysteines (e.g., Cys32-Cys41 in γD-crystallin) [132] is detrimental, as such crosslinking necessitates major protein misfolding and consequent aggregation (e.g., via exposure of hydrophobic residues, dissociation of β strands) (reviewed in [133]).
Due to the long-lived nature of crystallin proteins, the eye lens has evolved a sophisticated antioxidant defense system with unusually high levels of cellular reductants such as GSH that are responsible for balancing the lens redox state [134,135]. An active GSH redox cycle exists in the lens epithelium and superficial lens cortex, whereby GSH detoxifies potentially harmful oxidants, such as dehydroascorbic acid and hydrogen peroxide [114,136]. Aside from the detoxification of hydrogen peroxide, GSH also functions as a key hydroxyl radical scavenger in LECs [137]. In the central lens nucleus, the levels of GSH are particularly low and even more so with increasing age. A developing diffusion barrier near the periphery of the lens nucleus [138], together with little to no regeneration of GSH, makes the lens nucleus especially vulnerable to oxidative stress and subsequent cataract formation. This susceptibility was elegantly illustrated in the LEGSKO mouse model, where de novo GSH synthesis in the lens is abolished via knockout of γ-glutamyl-cysteine ligase [139,140,141]. LEGSKO mice show increased oxidation of crystallin and non-crystallin proteins (at methionine and/or cysteine residues), leading to severe ARN cataract by 9 months [139,140,141]. Within the old and GSH-depleted regions of the lens, the cysteine-rich and abundant γ-crystallins may help buffer against oxidative stress by serving as redox sinks [142,143,144,145]. However, dynamic disulfide exchange between γ-crystallins succeeds only in delaying the oxidative protein aggregation cascade [143,146]. Eventually, with age, kinetically favorable yet relatively innocuous disulfides formed early in the lens are shuffled to more thermodynamically stable, yet deleterious, ones that have direr consequences for crystallin stability [133,143]. The development of small-molecule drugs capable of penetrating lens nuclei to restore the redox balance and/or stem aggregation remains the key challenge in the pharmacological prevention or treatment of ARN cataract [114,147,148,149].
Oxidative stress is also a key driver of the pathogenesis of fibrotic forms of cataract, including posterior subcapsular cataract (PSC) [150] and PCO. A key underlying mechanism of fibrotic cataracts involves epithelial–mesenchymal transition (EMT), whereby the once regular, cuboidal lens epithelial cells dramatically transform into spindle-shaped mesenchymal cells [151,152,153,154]. In PSC and PCO, the equatorial LECs undergo EMT and migrate posteriorly to form a fibrotic plaque that impairs vision. Transforming growth factor-beta 2 (TGFβ2) is a classic inducer of EMT in the lens, resulting in the enhanced migration, contraction, and secretion of extracellular matrix proteins [151,155,156]. The induction of oxidative stress through hydrogen peroxide-induced lens EMT is mediated by the enhanced activation of the TGFβ/Smad and Wnt/β-catenin pathways [157]. Two antioxidants, GSH and catalase, have been shown to potently block TGFβ-induced cataract in cultured rat lenses and lens epithelial explants [158]. The depletion of GSH in the lens leads to increased EMT markers, including vimentin, α-SMA, fibronectin, and collagen type I [159]. ROS-induced extracellular vesicles have been shown to enhance EMT of LECs, which can be suppressed by the addition of diphenyleneiodonium, an NADPH oxidase inhibitor [160]. The lens-specific knockout of glutamate-cysteine ligase catalytic subunit (Gclc), which encodes the rate-limiting enzyme in GSH synthesis, resulted in elevated oxidative stress, inflammation, and the robust upregulation of cytokines [161]. The professional ROS producer, NADPH oxidase 4 (Nox4), has been shown to be upregulated in response to the TGFβ2 stimulation of rat lens epithelial explants, leading to EMT [162]. The blockade of Nox4 using a specific NADPH oxidase inhibitor reduced the TGFβ-induced upregulation of the gene expression of α-SMA, collagen 1a, and fibronectin [163], further emphasizing the key role of oxidative stress in driving lens EMT in the pathogenesis of fibrotic cataract.
5. Retina
The retina acts as the “sensor” of the eye, where the light is focused to interpret visual information. It is a highly organized tissue composed of nine histological layers comprising 50 different cell types, all resting on a pigmented epithelium. The retinal layers consist of photoreceptors (PRs), the external limiting membrane (ELM), the outer nuclear layer (ONL), the outer plexiform layer (OPL), the inner nuclear layer (INL), the inner plexiform layer (IPL), the ganglion cell layer (GCL), the nerve fiber layer (NFL), and the internal limiting membrane (ILM) [164,165].
The PRs in the outermost layer of the retina are responsible for transforming light energy into nerve impulses. There are approximately 100 million PRs distributed throughout the human retina, making them one of the most abundant types of neurons in the body, highlighting the herculean nature of image capture and processing [166,167]. The transformation of light energy by the photoreceptors is dependent on the alteration of visual pigments contained within the rods and cones [168]. These pigments consist of a Vitamin A-derived aldehyde, known as retinal, bound to large protein moieties, called opsins, which differ depending on the type of photoreceptor [164]. Light isomerizes retinal 11-cis to all-trans, releasing retinal from the protein moiety, a chemical sequence that promotes the transient excitation of the photoreceptor that propagates along its axon to stimulate second-order neurons, notably bipolar and amacrine cells [169]. These cells synapse onto retinal ganglion cells (RGCs) in the INL, whose axons coalesce to form the NFL and optic nerve, terminating at the lateral geniculate nucleus, which is connected by a second set of neurons to the visual cortex of the brain [170,171]. The integrity of the PR layer is heavily influenced by the retinal pigment epithelium (RPE), which consists of cells with sheet-like microvilli that interact with the outer PR segments extending from the outer retinal surface [172,173]. The RPE is essential in maintaining retinal homeostasis by facilitating the directional transport of nutrients and the clearance of photooxidized outer segment membranes [173]. Additionally, the RPE physically interacts with the outer retinal blood supply, known as the choroid, forming a structure known as the outer blood–retinal barrier (BRB). The choroid consists of five histological layers: Bruch’s membrane, the choriocapillaris, Sattler’s layer, Haller’s layer, and the suprachoroid [174].
The retina contains three major types of glial cells: microglia and two types of macroglia, astrocytes and Müller cells. Microglia are the primary innate immune cells of the retina. Suggested to be a heterogeneous population, they are in the GCL, in both plexiform layers, and around the vasculature as different subclasses [175,176]. Microglia constantly engage in the surveillance of surrounding neural tissue and contribute to the host defense against microorganisms, initiating inflammatory responses and promoting tissue repair in close association with macroglia and blood-borne immune cells [177,178]. Other roles for microglia include supporting vascular growth, the formation of neural connections, and neuronal apoptosis [179,180,181]. Given this association, microglial activation is thought to contribute to the progression of retinal degenerative diseases [182]. Astrocytes are primarily localized to the NFL and GCL and also exist as a heterogeneous population of three morphological subclasses interacting with nerve fibers, blood vessels, or the space between [183,184,185]. However, they are most well known for their role in supporting the retinal vasculature by ensheathing blood vessels, secreting trophic factors such as vascular endothelial growth factor (VEGF), and maintaining the integrity of the blood–retinal barrier [186,187].
Müller cells, the most common type of glial cell in the retina, extend radially across the entire retina to support cells in both the ILM and OLM [188]. Due to this orientation, Müller glia contact virtually every other retinal cell type, structurally minimize intraretinal light scattering, and help light focus onto the photoreceptors [189]. Müller cells provide critical homeostatic and trophic support to both the retinal vasculature and neuronal layers [190]. Among their many roles, Müller cells recycle neurotransmitters to prevent excitotoxicity, provide the spatial buffering of ions, reabsorb fluid to prevent edema, participate in the retinoid (visual) cycle, and regulate nutrient levels. In addition, Müller cells release a variety of neurotrophic and angiogenic growth factors and cytokines, as well as critical antioxidants such as GSH [191]. Müller cells are uniquely sensitive to metabolic perturbations and can become dysfunctional as they try to counter-regulate them [192,193]. This phenomenon is referred to as gliosis and is characterized by morphological and functional changes in the Müller glia. These changes manifest, in part, as alterations in gene and protein expression that contribute to retinal inflammation, microvascular defects, and neuronal dysfunction [192]. Prolonged or extensive damage can also induce metabolic memory, causing Müller cells to remain in a state of reactive gliosis even after the deleterious stimulus has dissipated.
5.1. Oxidative Stress and Retinal Vasculature
The retina is one of the most highly metabolically active tissues in the body and requires significant levels of oxygen and nutrients to maintain normal visual function. They are supplied by two key vascular networks: the central retinal artery system and the choriocapillaris [194]. Vessels extending from the central retinal arteries supply the inner half of the retina, branching into superficial, intermediate, and deep capillary layers after entering the eye via the center of the optic nerve. The choriocapillaris, directly adjacent to Bruch’s membrane, perfuses the outer retina and nourishes the RPE and photoreceptors [195]. Multiple cell types have been implicated in the development and maintenance of the retinal vasculature. RGCs are crucial contributors to maintaining the integrity of these vessels through the secretion of angiogenic factors and interact closely with them as the GCL is perfused by the superficial and intermediate vascular plexuses [196,197,198]. Pericytes are mural cells associated with the microcirculation that wrap around the endothelial cells that line systemic capillaries. Located in the basement membrane, they interact with the endothelium through both physical contacts that penetrate the basal lamina and via amacrine signaling. In the retina, they help support the vasculature, blood flow, and the BRB and respond to proangiogenic factors secreted by other cell types, such as RGCs. Combined with the metabolic and homeostatic support provided by glial cells, these cells coalesce to form the neurovascular unit to regulate retinal blood flow through vasodilation and vasoconstriction [199,200,201,202].
Given the high metabolic demand of the retina, its energy requirements are primarily met through oxidative and glycolytic metabolism with the input of additional nutrients by the retinal vasculature [203,204]. This can result in ROS/RNS production, which are tightly controlled by retinal glia and play important roles as secondary messengers in endothelial features [205,206]. Yet, under pathological conditions, these reactive species can accumulate, impacting the neurovascular unit and ultimately causing vascular disease [199,207]. For example, ROS accumulation causes an imbalance in nitric oxide metabolism, which impairs the response of vascular endothelial and smooth muscle cells to inflammation and changes in blood flow. Other consequences include increases in vascular permeability and retinal endothelial cell apoptosis [208,209]. These can occur as a result of a variety of pathological conditions and contribute to their overall progression while further exacerbating ROS production. The following sections discuss some of the more severe retinal diseases heavily associated with oxidative stress.
5.2. Oxidative Stress during Age-Related Macular Degeneration
Age-related macular degeneration (AMD), a retinal neurodegenerative disorder, is a common cause of irreversible central visual impairment in the aging population [210,211]. This ocular disease causes damage to the macular region of the retina, an oval-shaped, yellow area located in the central retina that facilitates high visual acuity and color vision due to the presence of the highest density of cone photoreceptors [212]. AMD is associated with a multifactorial etiology, with factors including increased age, female sex, obesity, genetics, a high-fat diet, and smoking [213]. There are two major forms of AMD: (i) dry AMD, which accounts for 90% of diagnosed AMD cases and is characterized by the formation of drusen between Bruch’s membrane and the retinal pigment epithelium (RPE), as well as the RPE and photoreceptor degeneration [214], and (ii) wet AMD, which is characterized by choroidal neovascularization (CNV) and involves the formation of new and leaky blood vessels driven by VEGF secretion, leading to macular edema, hemorrhage, and fibrous tissue proliferation [215,216].
The retinal microenvironment is prone to oxidative damage due to high oxygen consumption [217], exposure to visible light (400–700 nm), and the presence of high levels of polyunsaturated fatty acids and photosensitive molecules, including, lipofuscin and rhodopsin. With age, the decreased antioxidant capacity further accentuates the oxidative components, creating a harsh retinal microenvironment [218]. These age-related oxidative changes are central to AMD pathogenesis [219] (Figure 2), with increased ROS causing damage to cellular lipids, proteins, and DNA and impairing retinal function [220]. Previous studies have documented the presence of systemic oxidative stress in patients with AMD in comparison to non-AMD cohorts, indicated by the upregulation of MDA, 8-OHdG, and protein carbonyls [221]. Additionally, reduced levels of nitric oxide synthase (NOS) isoforms—neuronal (nNOS), inducible (iNOS), and endothelial (eNOS)—play an important role in AMD pathogenesis [222]. On the other hand, at high concentrations, NO is known to react with superoxide anions (O2·−) to generate peroxynitrite (ONNO−), which has the potential to accumulate protein aggregates between the RPE and photoreceptors, leading to their degeneration [223,224].
It has been documented that RPE cells derived from AMD patients lose their capacity to upregulate SOD expression upon oxidative stress exposure, thus leading to increased ROS accumulation compared to non-AMD RPE cells [225]. Moreover, mice deficient in SOD1 and SOD2 exhibit elevated levels of ROS, along with the development of specific features of AMD pathology, such as drusen formation, Bruch’s membrane thickening, and choroidal neovascularization [226,227]. Another antioxidant that plays an important role in the antioxidant response and retinal detoxification is nuclear factor erythroid 2-related factor 2 (NFR2). Downregulation of the NFR2 gene has been shown to increase retinal degeneration and lead to the development of an AMD-like phenotype with dysregulated autophagy, thus indicating a link between inflammation and oxidative stress [228]. A known Nrf2 activator, dimethyl fumarate, has been shown to suppress inflammation and metabolic dysfunction of the RPE [229] as well as blue-light-induced oxidative damage via the Nrf2 pathway [230]. Further, the accumulation of advanced glycation end products (AGEs) has been observed in the macular drusen of AMD patients, which are linked to enhanced inflammation, oxidative stress and vascular dysfunction [231,232]. One of the potential biomarkers in determining AMD susceptibility is carboxyethylpyrrole (CEP). Proteomic analyses of AMD patients have indicated significantly elevated levels of carboxyethylpyrrole protein adducts in their drusen [233]. CEPs are generated by the oxidation of the polyunsaturated fatty acid docosahexaenoic acid (DHA) in the outer segments of photoreceptors, making it a robust oxidative stress marker for AMD patients [234].
The retinal absorption of ultraviolet rays causes photochemical damage to mitochondria in the RPE, leading to increased ROS generation [235,236]. Specifically, blue light has been associated with significant photooxidative stress and ROS generation [237,238]. Light-induced damage is dependent on various chromophores, among which flavins and porphyrins have been identified as possible chromophores responsible for mitochondrial damage induced by blue light [239,240]. Moreover, the macular carotenoid lutein has antioxidant properties but also prooxidant properties at high concentrations [241]. Blue-light-mediated oxidative stress is proposed to occur in the outer photoreceptor segments through the NOX family of enzymes [242]. NOX2 and NOX4 increase ROS levels in photoreceptors irradiated with blue light, which has been shown to decrease with apocynin, a NOX inhibitor [242].
Another important risk factor for AMD is cigarette smoking. Studies have documented a 2–3-fold elevated risk of developing AMD in smokers compared to non-smokers [243]. Cigarette smoking has the ability to generate ROS and increase oxidative stress by elevating serum lipid peroxidation [244] and reducing the levels of antioxidants [245]. Additionally, it increases the levels of proinflammatory cytokines, including C-reactive pprotein (CRP) [246], which is associated with an increased risk of AMD. Notably, in vascular smooth muscle cells, CRP is known to exhibit prooxidative effects, triggering ROS accumulation and subsequent apoptosis [247] and promoting the adhesion of monocytes to endothelial cells through NOX-mediated oxidative stress [248]. Recently, studies have documented elevated levels of interleukin 1 beta (IL-1β), TNFα, and iNOS in mouse retinas following exposure to electronic cigarette vapor [249]. Furthermore, smoking is correlated with genetic susceptibility to AMD. For instance, the complement factor H Y402H polymorphism along with smoking has been associated with AMD [250].
In addition to environmental stressors and exogenous ROS, intracellular defects, such as dysregulated autophagy, have also been implicated in AMD progression. The impairment of the autophagic pathway leads to the decreased clearance of damaged organelles and proteins that can exacerbate oxidative stress and contribute to AMD pathogenesis [251]. In turn, the promotion of autophagy through the downregulation of hyperactive mTORC1 signaling by upregulating calcium and integrin binding protein 2 (CIB2) and the activation of the proliferator-activated receptor gamma coactivator 1 (PGC-1) [252] and AMPK-mTOR [253] signaling pathways have shown significant potential as novel therapies for dry and wet AMD [254,255]. Mitochondria play an important role in ROS generation since the ROS produced in the ETC can cause damage to cellular components. Previous studies have reported elevated levels of mtDNA and decreased DNA repair efficacy with the progression of AMD [255]. The downregulation of two key regulators of mitochondrial biogenesis and antioxidant production (nuclear factor erythroid 2-related factor 2 (NFE2L2) and PGC-1α) cause a disturbance in mitochondrial autophagy, known as mitophagy [256]. Previous reports have documented a decrease in mitophagy along with the accumulation of damaged mtDNA during AMD [257,258]. Along with this, the accumulation of iron in the retina has been observed in AMD. Excess iron is associated with ROS generation through the Fenton reaction [259]. In hereditary disorders such as hemochromatosis, which involves systemic iron overload, iron accumulation has been observed in the retina in spite of an intact blood–retinal barrier (BRB), resulting in clinical signs of AMD [260,261]. Iron-mediated oxidative stress has been reported in a sodium iodate-induced dry AMD model, indicating the significant upregulation of ferritin and impaired cathepsin D synthesis and maturation, resulting in iron-loaded ferritin accumulation [262].
At present, anti-VEGF therapy is approved for wet AMD. Various anti-VEGF agents, including aflibercept [263,264], bevacizumab, ranibizumab [265266267], and more recently, brolucizumab [268,269], have been used for the treatment of wet AMD. However, therapeutics for dry AMD remain limited. Numerous agents have been evaluated in the past for dry AMD treatment, including anti-inflammatory agents [270], immunomodulators, and antineoplastic agents [271]. Recently, a randomized phase II trial documented the efficacy of a complement 3 inhibitor, pegcetacoplan, in significantly reducing the progression of geographic atrophy in AMD patients [272].
Studies have supported the use of antioxidants in oxidative-stress-induced AMD models. Antioxidants such as zinc, resveratrol, and carotenoids have been proposed for dry AMD treatment [273,274]. Based on the Age-Related Eye Disease Study (AREDS), oral antioxidants are effective supplements for slowing AMD progression. Daily supplementation with select antioxidants, such as Vitamin C, Vitamin E, and beta-carotene along with zinc, was found to reduce the risk of AMD [275]. However, the follow-up clinical study, called the Age-Related Eye Disease Study 2 (AREDS2), testing the effects of further supplementation with omega-3-fatty acids DHA and EPA, along with carotenoids, lutein, and zeaxanthin, indicated no further reduction in AMD progression [276]. Supplementation with compounds such as Vitamin E, which has the ability to inhibit lipid peroxidation [277], has also shown protective properties in the RPE. Alpha-tocopherol transfer protein (α-TTP) has been associated with the maintenance of serum Vitamin E levels. The deletion of the α-TTP gene has been linked with retinal degeneration and the loss of photoreceptors. On the other hand, supplementation with Vitamin E was shown to suppress lipid peroxidation in a mouse model of neuronal degeneration [278]. While these collective findings suggest that supplementation with select antioxidants may delay AMD progression, further clinical trials are needed to establish therapeutic efficacy in larger, diverse populations and to develop preventative interventions to reduce retinal degeneration with age.
5.3. Oxidative Stress and Proliferative Vitreoretinopathy
Proliferative vitreoretinopathy (PVR) is a complication of rhegmatogenous retinal detachment surgery or severe ocular trauma, including intraocular foreign bodies, perforation, penetrating injury, contusion, or globe rupture [279]. Clinically, PVR is characterized by the formation of contractile fibrotic membranes on the epiretinal, intraretinal, and/or subretinal surfaces [280]. The rate of PVR is approximately 5–10% of patients undergoing retinal detachment surgery [281], and of these patients, less than 25% will achieve a visual acuity of 20/200 or better [282]. The only treatment for PVR is through the surgical excision of the fibrotic membrane [280]. The retinal cells identified in patient PVR membrane specimens predominantly comprise RPE but also glial cells (primarily Müller cells) and inflammatory cells (lymphocytes and macrophages). Interactions between these cells with a myriad of growth factors and cytokines derived from vitreous contact and breach of the blood–retinal barrier trigger a cascade of downstream cellular processes, including EMT, chemotaxis, proliferation, excessive extracellular matrix deposition, and cellular migration, leading to the formation of contractile fibrotic membranes and the overt loss of the retinal architecture [280].
Alterations in redox homeostasis have been implicated in the pathogenesis of PVR. Verdejo et al. (1999) examined the vitreous of patients undergoing vitrectomy for PVR, proliferative diabetic retinopathy, rhegmatogenous retinal detachment, macular hole, or epiretinal membrane [283]. Increased free radial formation and reduced antioxidant activity (SOD and catalase levels) were observed in the vitreous of PVR patients [283]. An increase in oxidative stress, proportional to disease severity, has been found in the vitreous of patients undergoing surgery for rhegmatogenous retinal detachment compared to macular hole [284,285]. Intriguingly, it has been observed that the retina may launch a protective response to injury by increasing its antioxidant defense system. Increased levels of the radical scavenger α1-microglobulin were found in patients with rhegmatogenous retinal detachment compared to macular hole [284]. Further support for launching a protective antioxidant response was shown by Pietras-Baczewska et al. (2021), where increases in two antioxidative enzymes, SOD and glutathione reductase, were detected in the vitreous of patients with PVR compared to patients with macular hole or epiretinal membrane [286]. Regardless of the discrepancies in the precise changes in oxidative stress and antioxidant levels, it is clear that an imbalance of ROS and antioxidant defenses occurs during PVR. This may trigger subsequent damage, including lipid peroxidation, cellular degeneration, and the release of diverse regulatory molecules and cytokines that drive the further progression of the disease. Reduced GSH concentrations in vitreous and blood samples of PVR patients have been detected [287]. Oxidative tissue damage through lipid peroxidation, as measured by thiobarbituric acid-reactive substances, has been identified in epiretinal membranes isolated from patients with PVR [288].
EMT of the RPE is a predominant process driving PVR pathogenesis [289]. During EMT, the regular and polarized RPE cells transform into motile, matrix-producing mesenchymal cells that secrete and deposit excessive extracellular matrix [290,291]. These transdifferentiated myofibroblasts migrate along the subretinal plane and enter the epiretinal plane by migrating through retinal breaks [280]. TGFβ is the primary growth factor involved in the induction of EMT of the RPE and is present at high concentrations in vitreous isolated from PVR patients [292]. Oxidative stress was found to enhance TGFβ-induced EMT through the upregulation of the inflammatory cytokine chemokine ligand 1 (CXCL1) in the ARPE-19 cell line [293]. In this study, hydrogen peroxide induced the upregulation of CXCL1 which, in turn, upregulated mesenchymal markers such as α-SMA and fibronectin in the RPE [293], highlighting a synergistic effect with inflammation, oxidative stress, and EMT. While RPE cells physiologically express NOX1, NOX2, and NOX4, dysregulation of NOX4 signaling has been shown to augment EMT [294,295,296]. VAS2870, a novel NAPDH oxidase inhibitor, blocked the TGFβ-induced EMT of the RPE [297]. The administration of an antioxidant, αB crystallin peptide, significantly reduced vimentin, fibronectin, and CTGF levels in a dispase-induced PVR mouse model and, in doing so, improved the mitochondrial function of the RPE [298]. Other antioxidants, such as quercetin [299], ZLN005 [300], and artesunate [301], have shown efficacy in blocking the TGFβ-induced EMT of the RPE, further highlighting a key role for oxidative stress in PVR pathogenesis.
5.4. Oxidative Stress and Diabetic Retinopathy
Diabetic retinopathy (DR) is the leading cause of acquired blindness in working-age adults in Western countries, with approximately 90% of diabetic individuals experiencing vision-threatening complications within 25 years of their initial diagnosis [302]. In the clinic, DR is primarily characterized by vascular abnormalities that can be detected by current diagnostic techniques. These include microcirculatory complications that affect the capillaries in the eye, such as microaneurysms, hemorrhaging, and angiogenesis, which are all associated with the more severe, late-stage aspects of DR that can lead to retinal detachment and blindness [303].
In addition to vascular abnormalities, the neurosensory retina is altered in diabetes. The neurosensory retina generates vision but is transparent and largely undetectable by standard clinical examination. However, most retinal neurons and glial cells are altered prior to the development of microvascular lesions and are progressively impaired with worsening retinopathy. These alterations include biochemical defects, such as impaired control of glutamate metabolism (the major neurotransmitter) [304], as well as the loss of synaptic activity and dendrites [305], apoptosis of neurons primarily in the ganglion cell and inner nuclear layers [306,307], and the activation of microglial cells that may protect the inner retina from injury and contribute to the inflammatory response [308].
With improvements in understanding the pathogenesis of diabetes over the past several decades, it has been well accepted that most complications associated with diabetes are linked to the severity of hyperglycemia. Two of the most comprehensive studies exploring this relationship, The Diabetes Complications and Control Trial (DCCT) and the UK Prospective Diabetes Study (UKPDS), report that hyperglycemia is the dominant contributor to the manifestation of DR in both type 1 and type 2 diabetes [309,310]. Both reports also indicate a significant reduction in DR development with intensive glycemic control. Four key pathways have been implicated in the effects of hyperglycemia-induced tissue damage: the polyol pathway, AGE pathway, hexosamine biosynthetic pathway, and protein kinase C pathway.
A unifying theory for the pathobiology of diabetic complications suggests that the activation of each of these pathways is linked to the excessive production of ROS [311]. While acute bursts of ROS are important in normal physiological signaling through their role as secondary messengers, prolonged exposure can result in damage to macromolecules [312]. This includes proteins, lipids, and nucleic acids, whose modification by ROS can impede normal physiological function [313]. Thus, retinal cells must maintain the tight regulation of ROS by balancing their production and clearance [314]. Sources of ROS within the retina include NADP(H) oxidases, xanthine oxidases, and retinal excitation by light [315,316,317,318]. Yet, the dominant source of retinal ROS is the mitochondrial ETC [319,320]. Photoreceptors and the RPE are two retinal cell types that have been found to contain a high mitochondrial density. Previous work suggests that hyperglycemia increases ΔΨm, which leads to excess superoxide production through enhanced glycolytic flux and a surplus of electron carriers generated by the TCA cycle [321,322,323,324]. The carriers enter through ETC complexes I and II, resulting in a ΔΨm that overloads complex III and generates superoxide [325]. Additional evidence substantiates this by describing a multicomponent feedback loop that continues to drive this harmful production of ROS [326,327]. Non-enzymatic antioxidants such as β-carotene, Vitamin C, and Vitamin E are all negatively impacted by hyperglycemia [328]. Enzymatic factors, which include proteins such as SOD, catalase, and enzymes involved in the synthesis of GSH, are also negatively influenced by hyperglycemia [329,330,331,332].
Increases in reactive nitrogen species (RNS), including the nitroxyl anion, the nitrosonium cation, and peroxynitrite, are also coincident with elevated ROS. It has been reported that the increased activation and expression of eNOS is commonly seen in models of type 1 and type 2 diabetes and can result in elevated levels of NOS that can ultimately cause neurological and vascular complications [333,334,335,336]. While nitric oxide is also important in cellular signaling, it can react with O22− to form a peroxynitrite radical, which is thought to have cytotoxic effects under hyperglycemic conditions [337,338,339].
In addition to ROS and RNS, studies have demonstrated that diabetes-induced hyperglycemia causes a failure to upregulate factors associated with the antioxidant response [340,341,342,343]. Notably, patients diagnosed with type 1 and type 2 diabetes have been shown to exhibit a significant reduction in antioxidants [344,345,346]. This exacerbates the accumulation of harmful reactive species, creating the imbalance known as oxidative stress, contributing to the hyperglycemia-induced tissue damage that drives DR pathogenesis (Figure 3).
Hyperglycemia compromises the antioxidant response [347,348,349], owing, in part, to enhanced degradation due to the dysregulated diabetic metabolic microenvironment [343]. The Nrf2 gene battery consists of more than 200 genes associated with not only the redox balance but also processes such as inflammation and proteostasis [350]. Thus, its augmentation has been of great interest in developing therapies to combat DR.
The retina contains several different cell types that exhibit high metabolic activity through the consumption of large quantities of glucose and oxygen [351]. It also possesses some of the highest concentrations of PUFAs among all the tissues in the body, making it sensitive to lipid peroxidation [352]. As a result, any changes in the redox balance due to diabetes-induced hyperglycemia are likely to alter retinal structure and function. Previous reports have demonstrated a clinical correlation between free radical formation and the development of DR. This has also been supported by several in vivo studies that report the accumulation of ROS [315,321,327,353,354,355] and RNS [356,357,358,359] in retinal tissues of diabetic rodents. The exposure of endothelial cells, pericytes, neurons, and Müller cells to hyperglycemic conditions has been shown to result in the accumulation of these free radicals and can progress to retinal cell apoptosis [327,329,360,361,362,363]. Müller cells, astrocytes, and photoreceptors have also been shown to be affected early in DR pathogenesis, resulting in elevated ROS/RNS levels that cause further damage to these retinal cells [343,364,365,366].
The suppression of the antioxidant defense has been reported in multiple DR models [343,367]. Pericytes isolated from the postmortem retinas of diabetic patients exhibited the downregulation of mRNAs encoding glutathione reductase and SOD upon exposure to hyperglycemic culture conditions when compared to those from non-diabetic patients [368]. In both postmortem samples and preclinical models from diabetic patients, retinal Nrf2 DNA-binding activity was found to be reduced [26,343,369]. In rodents, streptozotocin (STZ)-induced diabetes suppressed retinal GSH levels and increased lipid peroxidation [370,371]. In vitro studies exposing bovine pericytes to AGEs have also documented a decrease in intracellular catalase and SOD activities [372].
Antioxidant supplementation has become an avenue of interest for combatting DR pathogenesis by addressing earlier deficits in the neurosensory retina that are not targeted by current therapeutics, while also hindering progression to later stages of the disease. In diabetic rats, the administration of Vitamins C and E reduced the appearance of acellular capillaries and pericyte ghosts and halted DR progression in alloxan-induced diabetic rats [329]. In vitro treatment with α-lipoic acid and N-acetyl cysteine, the precursor of the amino acid cysteine required for GSH synthesis, inhibited oxidative-stress-induced damage in retinal endothelial cells, pericytes, and neuronal precursor cells [327,373]. The administration of α-lipoic acid also prevented retinal capillary cell death and the development of microvascular defects in diabetic rats [329]. Dietary N-acetyl cysteine supplementation attenuated the development of vascular pathology in the retina of STZ-treated diabetic rats and has been shown to prevent retinal cell death and contrast sensitivity deficits in STZ-treated diabetic mice [327]. Unfortunately, data obtained from multiple clinical trials showed no protective effect of β-carotene or Vitamin C and E supplementation in DR patients [374,375,376]. However, this does not discount the potential benefit of modulating ROS-generating or antioxidant defense pathways as therapeutic options in DR.
5.5. Oxidative Stress Mechanisms in Glaucoma
Glaucoma is the second leading cause of blindness, with estimates of 80 million glaucoma patients expected to be diagnosed by 2040 [377]. Glaucoma patients are characterized by a progressive loss of RGCs and their axons and subsequent visual field loss. Elevated intraocular pressure (IOP) is a well-known risk factor for glaucoma and is a diagnostic clinical sign of primary open-angle glaucoma (POAG) [378]. Elevated IOP and dysregulated blood flow in the retina may cause initial damage to the optic nerve. Current glaucoma therapies merely slow disease progression, with no treatment available for reversing glaucomatous neurodegeneration. Thus, exploration of the underlying mechanisms is critical for designing novel treatments for glaucoma. Glaucoma is a multifactorial retinal degenerative disease that manifests as different subtypes, such as POAG and primary angle closure glaucoma (PACG). POAG is the more common form of glaucoma compared to PACG [377]. In normal-tension glaucoma (NTG), patients present with glaucomatous optic neuropathy but exhibit IOP in the normal range, that is, between 11 and 21 mmHg. NTG is more common in Asian populations (52–92%) [379] and presents in 30–40% of Caucasian patients with POAG [380].
The mechanisms of the progressive degeneration of RGCs and their axons in glaucoma have been attributed to various factors, such as the elevation of IOP, the activation of immune cells, altered trabecular meshwork (TM) cells, and dysregulated gut microbiota. Oxidative stress is one of the candidates leading to POAG and RGC degeneration. The antioxidant capacity in patients with POAG is reduced by over 60%, suggesting that these patients might be more susceptible to oxidative stress of the TM and retinal tissues [381,382].
The retina is the most energy- and oxygen-demanding tissue in the body and exhibits intense mitochondrial activity [383]. RGCs consume 5-fold more ATP than photoreceptors in the dark [384]. The imbalance of ROS and antioxidant enzymes are critical to the health of retinal neurons. Excess and unstable ROS may react with lipid membranes, proteins, and nucleic acids, leading to RGC degeneration and apoptosis [385,386]. Elevated IOP may affect the drainage facility of TM cells and reduce blood flow [387], imposing stress on retinal structures, especially the anterior segment of the optic nerve. This interferes with axonal transport, leading to RGC degeneration. Oxidative stress is considered one of the major drivers of damage to TM cells in glaucoma [388]. TM cells could be damaged by elevated ROS levels and lead to alterations in the drainage facility of the TM [389].
In addition to ROS production, oxidative stress could affect DNA modifications [390] through alkylation, deamidation, and oxidation, eventually leading to cell apoptosis [12,391]. These DNA modifications can be repaired by base excision repair mechanisms [390]. An association between base excision repair gene polymorphisms and a higher risk of POAG in Polish populations has been reported [392]. Clinical studies have shown the upregulation of a DNA oxidation marker, 8-OHdG, in the blood, aqueous humor, and TM from POAG patients [393]. Changes in 8-OHdG in the TM have been correlated with visual field defects [393]. As further support for the impact of oxidative stress on DNA in glaucoma, a negative correlation between base excision repair gene expression (PARP1 and OGG1) and the 8-OHdG level has been detected in the plasma of glaucoma patients [394]. The chronic exposure of TM cells to oxidative stress leads to reduced cell viability and increased cell senescence [395] via the upregulation of cyclin-dependent kinase inhibitors p16 and p21 [396]. Senescent cells are the active source of proinflammatory cytokines, chemokines, and extracellular-matrix-degrading proteins that may promote a toxic environment for RGCs [397,398].
The response to oxidative stress is mainly regulated by the mitochondria of RGCs and their axons [399]. In a study using the DBA/2J mouse model of congenital glaucoma, mitochondrial dysfunction was found to be the key driver of early neuronal dysfunction before RGC degeneration [400]. NAD is a highly conserved coenzyme central to metabolism and serves as an important redox cofactor, providing electrons to complex I of the ETC for ATP production. Nicotinamide treatment was shown to protect against RGC loss in DBA/2J mice [401], which is further supported by observations of NAD deficiency in POAG patients [402]. Mitochondrial dysfunction releases ATP and ROS, activating the NLRP3 inflammasome and releasing potent inflammatory signals [403]. Excessive ROS can be neutralized by antioxidant defense mechanisms or enzymes, including SOD, glutathione peroxidase, catalase, and glutathione reductase [404]. Accumulating evidence supports redox dysregulation in glaucoma patients, revealing an imbalance in ROS and antioxidant levels. Intriguingly, levels of individual antioxidant enzymes do not always show reduced expression in glaucoma patients. For example, the expression of SOD1 was downregulated in blood samples from POAG patients, but SOD2 expression was upregulated [405]. The knockout of antioxidant-defense-system-related genes (SOD1, SOD2, NRF2) in animal models typically results in progressive neuro- and retinal degeneration, with loss of RGCs, decreases in ERG waves, and increases in cleaved-caspase-3-positive apoptotic cells in the retina and varying degrees of disease progression with age [227,228,406]. However, none of these models investigated the effect of elevated IOP, so the conclusions derived from these studies are for general retinal neurodegeneration.
NOX enzymes are also key contributors to oxidative stress. Excessive oxidative stress may generate free radicals, such as the superoxide anion, hydroxyl radical, and nitric oxide. NOX, xanthine oxidase, uncoupled NO synthase, and defects in the ETC lead to excessive ROS generation [407,408], especially the latter one [383,384,409]. The catalytic unit of NOX has seven isoforms, NOX1–5 and DUOX1–2. NOX expression has been implicated in the pathogenesis of various animal models of glaucoma [410]. NOX1 and NOX2 generate superoxide, and NOX4 generates hydrogen peroxide [411]. Disruption of the NOX system in animal models, however, primarily leads to photoreceptor degeneration [412], whereas the primary feature of POAG is RGC loss.
RNS such as uncontrolled nitric oxide production (NO, a well-known vasodilator) can induce oxidative stress, leading to apoptosis [413]. The upregulation of iNOS and the downregulation of calcium/calmodulin-dependent NOS expression were correlated with visual defects in POAG patients [414]. Endothelin-1 (ET-1), a well-known vasoconstrictor, has been shown to increase in POAG patients [415,416,417]. Dysregulation of vasodilation and vasoconstriction could affect the RGC metabolic supply and increase oxidative stress, leading to RGC degeneration [418]. Clinical studies have shown that total biological or total reactive antioxidant levels are generally reduced in blood samples collected from glaucoma patients, correlating with glaucoma severity [419,420,421,422]. Furthermore, increased MDA levels, a well-known secondary product of lipid peroxidation [423], in the blood and aqueous humor [424], and reduced antioxidant levels [424,425] have been detected in glaucoma patients.
Microglial activation is an early pathophysiological change that has been shown to precede RGC loss in both glaucoma patients and the glaucomatous DBA/2J mouse model [426]. Transcriptome studies in microglia in the optic nerve head indicate significant changes in metabolic, phagocytotic, inflammatory, and sensome-related genes [427]. The upregulation of the major proinflammatory cytokine TNF-α is well-known in glaucoma [428,429,430,431]. Both TNF-α and heat shock proteins (released from damaged, stressed, or dead cells) upregulate NOX activities and ROS production in microglia [432,433], which have been suggested to play a key role in the pathogenesis of glaucoma [434]. Activated proinflammatory microglia have been implicated in neurotoxicity and RGC phagocytosis and are considered a significant contributor to RGC loss in glaucoma [435]. In contrast, microglia depletion studies using CSF1R blockers show increased RGC loss and progressive astrocytosis using the microbead-induced glaucoma model [436], suggesting a protective role for microglia in glaucoma, highlighting the complexity of the involvement of microglia in glaucoma.
Several preclinical and clinical trials involving antioxidant substances were conducted in glaucoma animal models and clinical trials with promising results: astaxanthin (AST) [437], Vitamin B [401], resveratrol, ascorbyl laurate [438], Ginkgo biloba extract [439], coenzyme Q10 and Vitamin E [440], DHA [441], and many others demonstrate the ameliorative effects of antioxidants on visual functions, retinal blood flow, ERG function, and RGC loss, further establishing glaucoma as an oxidative-stress-mediated pathology. However, while encouraging, none of these antioxidant treatments can fully arrest glaucoma progression.
The pathophysiology of glaucoma is a multifactorial neurodegenerative process affecting multiple cell types in the retina, optic nerve, and TM, leading to complex changes in the microenvironment of the eye. Further investigation of the contribution of various retinal cell types, including neurons and glial cells, and their crosstalk is required to further evaluate the potential of targeting oxidative stress to treat glaucoma or other forms of optic neuropathy. Despite the clear evidence of the prominent role of oxidative stress in the development and progression of POAG, the complexity of POAG pathogenesis has hindered the identification of the precise genes and pathways involved. It is also an obstacle to developing transgenic mouse models that fully recapitulate the pathophysiology of POAG. Further investigations using advanced technologies such as single-cell RNA sequencing open new possibilities for identifying the gene expression signatures of different cell types associated with POAG.
6. Targeting Oxidative Stress in Aging
While aging has long been considered a non-modifiable risk factor for many diseases, including those mentioned throughout this review, current trends advocate for the concept that aging itself is a preventable disease [442,443]. Moreover, the processes that underlie resilience and frailty appear to involve many of the same comparative indices that define biological age [444,445,446] and involve the mechanisms in place that promote cellular defenses against oxidative stress, as well as the maintenance of genomic integrity and optimal mitochondrial function [4]. Studies have shown that sera from young mice can promote tissue rejuvenation in older mice, suggesting that factors found in the circulation are important in aging and longevity [447,448]. Consistent with this idea, the activation of conserved adaptive stress responses appears to reduce the pathological effects of aging [449].
Therapeutic interventions to delay age-related degeneration via metabolic regulation have shown promising results in animal models with a focus on longevity as the endpoint [450,451] and are currently under investigation in human clinical trials [452]. An example of modulating bioenergetics to improve physical function has been shown in preclinical studies of the small molecule 1,1-dimethylbiguanide hydrochloride, commonly known as metformin, which is a treatment for type 2 diabetes mellitus and a potent metabolic regulator that promotes improved responsiveness to insulin and aids in stabilizing blood glucose levels [453]. The extended dietary intake of metformin in mice has been found to decrease the expression of markers associated with oxidative damage in the liver (e.g., lysine-4-hydroxinonenal and 8-iso-PGF2α), correlating with a ~6% increase in overall lifespan and lower mean body weight [450]. The secondary effects of metformin have been attributed to the activation of the metabolic regulator AMP-activated protein kinase and the potent antioxidant enzyme peroxiredoxin-2 [454]. Metformin has been proposed as a treatment for ocular conditions associated with mitochondrial dysfunction [455,456,457]. Retrospective studies have shown a modest trend of a reduced risk of AMD in subjects taking metformin, though confounding factors such as the presence of cardiovascular disease and diabetes have likely contributed to conflicting findings [458].
At the center of these mechanisms are mitochondrial function and the ubiquitous molecule NAD, a metabolite involved in major metabolic pathways (glycolysis and the TCA cycle, among others) and an important cofactor of the pro-reparative sirtuin enzymes. Preclinical studies have focused heavily on the mitochondrial isoform, SIRT3, an NAD+-dependent deacetylase important in the regulation of mitochondrial activity and strongly expressed in highly metabolically active organs, e.g., the heart, liver, kidney, smooth muscle, and brain [459]. Decreased SIRT3 expression in the brain has been associated with Alzheimer’s disease in both human [460] and animal tissues [461], though only small and selective differences in acetylation patterns in human postmortem tissues have been reported [462]. The total or partial loss of SIRT3 in mouse models appears to yield a dramatic phenotype and is associated with hyperacetylation, lower mitochondrial protein expression, and the increased expression of neuroinflammatory markers in the brain [463]. In contrast, the overexpression of SIRT3 in the hippocampi of aged mice appears to decrease neuroinflammation following surgery [464]. Like metformin, NAD supplementation has also been proposed as a small molecule capable of promoting favorable energy utilization and improving overall health [465]. These studies provide a degree of support for the hypothesis that metabolic regulation and mitochondrial integrity are important pathways involved in age-related conditions, which may be modifiable with therapeutic intervention. However, whether these mechanisms can be translated to a decrease in morbidities associated with age-related conditions of the eye remains unclear.
7. Conclusions and Future Directions
Redox homeostasis is a delicate balancing act of maintaining appropriate levels of antioxidant defense mechanisms and reactive oxidizing species (ROS and RNS). Any disruption of this balance leads to oxidative stress, which is a key pathogenic factor in several ocular diseases, as highlighted in this review. While excessive reactive oxidizing species are most definitely detrimental to cell function, the same is true of excessive antioxidants. Indeed, physiological levels of ROS and RNS are required for normal cellular function, as they act as second messengers in redox signaling [466]. Second messengers are generated at the instant of receptor activation and act specifically to relay signals to their effectors in a rapid and transient manner. For instance, the mitogen-activated protein kinase (MAPK) family of signaling molecules, including ERK, JNK, and p38, are activated through specific signal transduction cascades that can be activated by H2O2 [467]. Thus, antioxidant therapies must take into consideration this delicate redox balance and ensure that basal levels of reactive species are maintained for proper cellular functioning.
A myriad of different cell types exists in ocular tissues, each presenting different levels of mitochondrial activity and, thus, different oxidative stress susceptibilities and antioxidant defense mechanisms [468]. For example, mature lens fiber cells in the lens nucleus are devoid of organelles and thus possess no mitochondria and are metabolically quiescent. In contrast, the RPE and photoreceptors are packed with mitochondria to cater to their numerous metabolically demanding functions. While mature lens fiber cells are exposed to less ROS compared to the RPE, they must also depend on the slow, passive diffusion of antioxidants into the central lens nucleus, whereas RPE cells have active organelles to generate a robust antioxidant defense system.
A key unifying risk factor for oxidative stress in ocular diseases is aging. Increased reactive oxidizing species and weakened antioxidant defense systems are associated with increasing age, leading to genetic and epigenetic DNA mutations and subsequent disease. Mitochondria are particularly susceptible to the effects of aging. Metabolic changes and acquired deletions and mutations in mtDNA accumulate over time due to a low replication fidelity, a highly oxidative microenvironment, and the lack of protective histones present in mitochondria compared to the nucleus. These collective changes may contribute to the development of various conditions affecting both the anterior and posterior segments of the eye, as presented in this review (Table 2). Great strides have been made in the fields of oxidative stress, aging, and ocular disease. Continued efforts in this field will not only yield a better understanding of the molecular mechanisms underlying the pathogenesis of ocular diseases but also enable the identification of novel druggable redox targets and antioxidant therapies.
Author Contributions
Conceptualization, T.B.M. and D.Y.S.; methodology, D.Y.S., S.C., K.-S.C., A.L., W.P.M., D.C.T., M.Y. and T.B.M.; formal analysis, D.Y.S., S.C., K.-S.C., A.L., W.P.M., D.C.T., M.Y. and T.B.M.; investigation, D.Y.S., S.C., K.-S.C., A.L., W.P.M., D.C.T., M.Y. and T.B.M.; resources, D.Y.S., S.C., K.-S.C., A.L., W.P.M., D.C.T., M.Y. and T.B.M.; writing—D.Y.S., S.C., K.-S.C., A.L., W.P.M., D.C.T., M.Y. and T.B.M. review and editing—D.Y.S., S.C., K.-S.C., A.L., W.P.M., D.C.T., M.Y. and T.B.M.; All authors have read and agreed to the published version of the manuscript.
Funding
DYS is funded by the BrightFocus Foundation Postdoctoral Fellowship Program in Macular Degeneration Research (M2021010F). D.C.T. is supported by a National Eye Institute, National Institutes of Health, grant (R01EY030444).
Acknowledgments
Schematics were created using Biorender.com.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Niccoli, T.; Partridge, L. Ageing as a risk factor for disease. Curr. Biol. 2012, 22, R741–R752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, D.; LaBella, K.A.; DePinho, R.A. Telomeres: History, health, and hallmarks of aging. Cell 2021, 184, 306–322. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinnery, P.F.; Samuels, D.C.; Elson, J.; Turnbull, D.M. Accumulation of mitochondrial DNA mutations in ageing, cancer, and mitochondrial disease: Is there a common mechanism? Lancet 2002, 360, 1323–1325. [Google Scholar] [CrossRef]
- Marcelino, L.A.; Thilly, W.G. Mitochondrial mutagenesis in human cells and tissues. Mutat. Res. DNA Repair 1999, 434, 177–203. [Google Scholar] [CrossRef]
- Cortopassi, G.A.; Shibata, D.; Soong, N.W.; Arnheim, N. A pattern of accumulation of a somatic deletion of mitochondrial DNA in aging human tissues. Proc. Natl. Acad. Sci. USA 1992, 89, 7370–7374. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Pursell, Z.F.; Copeland, W.C.; Longley, M.J.; Kunkel, T.A.; Mathews, C.K. DNA precursor asymmetries in mammalian tissue mitochondria and possible contribution to mutagenesis through reduced replication fidelity. Proc. Natl. Acad. Sci. USA 2005, 102, 4990–4995. [Google Scholar] [CrossRef] [Green Version]
- Lodish, H.; Berk, A.; Kaiser, C.A.; Amom, A.; Ploegh, H.; Bretscher, A.; Kriefer, M.; Martin, K.C. Molecular Cell Biology, 8th ed.; Macmillan: New York, NY, USA, 2016. [Google Scholar]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, T.; Tsuchiya, S. Superoxide production from nonenzymatically glycated protein. FEBS Lett. 1988, 236, 406–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badwey, J.A.; Curnutte, J.T.; Karnovsky, M.L. Cis-Polyunsaturated fatty acids induce high levels of superoxide production by human neutrophils. J. Biol. Chem. 1981, 256, 12640–12643. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Kawanishi, S. Hydroxyl radical production and human DNA damage induced by ferric nitrilotriacetate and hydrogen peroxide. Cancer Res. 1987, 47, 6522–6527. [Google Scholar]
- Wagner, J.R.; Hu, C.-C.; Ames, B.N. Endogenous oxidative damage of deoxycytidine in DNA. Proc. Natl. Acad. Sci. USA 1992, 89, 3380–3384. [Google Scholar] [CrossRef] [Green Version]
- Boveris, A.; Oshino, N.; Chance, B. The cellular production of hydrogen peroxide. Biochem. J. 1972, 128, 617–630. [Google Scholar] [CrossRef]
- Sundaresan, M.; Yu, Z.-X.; Ferrans, V.J.; Irani, K.; Finkel, T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 1995, 270, 296–299. [Google Scholar] [CrossRef] [Green Version]
- Traverso, N.; Menini, S.; Maineri, E.P.; Patriarca, S.; Odetti, P.; Cottalasso, D.; Marinari, U.M.; Pronzato, M.A. Malondialdehyde, a lipoperoxidation-derived aldehyde, can bring about secondary oxidative damage to proteins. J. Gerontol. A Biol. Sci. Med. Sci. 2004, 59, B890–B895. [Google Scholar] [CrossRef] [Green Version]
- Refsgaard, H.H.; Tsai, L.; Stadtman, E.R. Modifications of proteins by polyunsaturated fatty acid peroxidation products. Proc. Natl. Acad. Sci. USA 2000, 97, 611–616. [Google Scholar] [CrossRef] [Green Version]
- Iles, K.E.; Liu, R.-M. Mechanisms of glutamate cysteine ligase (GCL) induction by 4-hydroxynonenal. Free Radic. Biol. Med. 2005, 38, 547–556. [Google Scholar] [CrossRef]
- Uchida, K.; Shiraishi, M.; Naito, Y.; Torii, Y.; Nakamura, Y.; Osawa, T. Activation of stress signaling pathways by the end product of lipid peroxidation: 4-hydroxy-2-nonenal is a potential inducer of intracellular peroxide production. J. Biol. Chem. 1999, 274, 2234–2242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Behndig, A.; Svensson, B.; Marklund, S.L.; Karlsson, K. Superoxide dismutase isoenzymes in the human eye. Investig. Ophthalmol. Vis. Sci. 1998, 39, 471–475. [Google Scholar]
- Deliyanti, D.; Alrashdi, S.F.; Tan, S.M.; Meyer, C.; Ward, K.W.; de Haan, J.B.; Wilkinson-Berka, J.L. Nrf2 Activation Is a Potential Therapeutic Approach to Attenuate Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2018, 59, 815–825. [Google Scholar] [CrossRef] [Green Version]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2017, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balboa, M.A.; Balsinde, J. Oxidative stress and arachidonic acid mobilization. Biochim. Biophys. Acta 2006, 1761, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Brash, A.R. Arachidonic acid as a bioactive molecule. J. Clin. Investig. 2001, 107, 1339–1345. [Google Scholar] [CrossRef] [Green Version]
- Cummings, B.S.; McHowat, J.; Schnellmann, R.G. Phospholipase A(2)s in cell injury and death. J. Pharmacol. Exp. Ther. 2000, 294, 793–799. [Google Scholar] [PubMed]
- Colston, J.T.; de la Rosa, S.D.; Strader, J.R.; Anderson, M.A.; Freeman, G.L. H2O2 activates Nox4 through PLA2-dependent arachidonic acid production in adult cardiac fibroblasts. FEBS Lett. 2005, 579, 2533–2540. [Google Scholar] [CrossRef] [Green Version]
- Sporn, P.H.; Marshall, T.M.; Peters-Golden, M. Hydrogen peroxide increases the availability of arachidonic acid for oxidative metabolism by inhibiting acylation into phospholipids in the alveolar macrophage. Am. J. Respir. Cell Mol. Biol. 1992, 7, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Cane, A.; Breton, M.; Koumanov, K.; Béréziat, G.; Colard, O. Oxidant-induced arachidonic acid release and impairment of fatty acid acylation in vascular smooth muscle cells. Am. J. Physiol. 1998, 274, C1040–C1046. [Google Scholar] [CrossRef] [PubMed]
- Matono, R.; Miyano, K.; Kiyohara, T.; Sumimoto, H. Arachidonic acid induces direct interaction of the p67(phox)-Rac complex with the phagocyte oxidase Nox2, leading to superoxide production. J. Biol. Chem. 2014, 289, 24874–24884. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.J.; Liang, C.L.; Li, G.M.; Yu, C.Y.; Yin, M. Neuroprotective effects of arachidonic acid against oxidative stress on rat hippocampal slices. Chem. Biol. Interact. 2006, 163, 207–217. [Google Scholar] [CrossRef]
- DelMonte, D.W.; Kim, T. Anatomy and physiology of the cornea. J. Cataract. Refract. Surg. 2011, 37, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Hassell, J.R.; Birk, D.E. The molecular basis of corneal transparency. Exp. Eye Res. 2010, 91, 326–335. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Hill, L.J.; Downie, L.E.; Chinnery, H.R. Neuroimmune crosstalk in the cornea: The role of immune cells in corneal nerve maintenance during homeostasis and inflammation. Prog. Retin. Eye Res. 2022, 91, 101105. [Google Scholar] [CrossRef]
- Alves, M.; Novaes, P.; Morraye Mde, A.; Reinach, P.S.; Rocha, E.M. Is dry eye an environmental disease? Arq. Bras. Oftalmol. 2014, 77, 193–200. [Google Scholar] [CrossRef] [Green Version]
- Seen, S.; Tong, L. Dry eye disease and oxidative stress. Acta Ophthalmol. 2018, 96, e412–e420. [Google Scholar] [CrossRef] [Green Version]
- Junqueira, V.B.; Barros, S.B.; Chan, S.S.; Rodrigues, L.; Giavarotti, L.; Abud, R.L.; Deucher, G.P. Aging and oxidative stress. Mol. Asp. Med. 2004, 25, 5–16. [Google Scholar] [CrossRef]
- Nakamura, S.; Shibuya, M.; Nakashima, H.; Hisamura, R.; Masuda, N.; Imagawa, T.; Uehara, M.; Tsubota, K. Involvement of oxidative stress on corneal epithelial alterations in a blink-suppressed dry eye. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1552–1558. [Google Scholar] [CrossRef] [PubMed]
- Wakamatsu, T.H.; Dogru, M.; Matsumoto, Y.; Kojima, T.; Kaido, M.; Ibrahim, O.M.; Sato, E.A.; Igarashi, A.; Ichihashi, Y.; Satake, Y.; et al. Evaluation of lipid oxidative stress status in Sjögren syndrome patients. Investig. Ophthalmol. Vis. Sci. 2013, 54, 201–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kojima, T.; Wakamatsu, T.H.; Dogru, M.; Ogawa, Y.; Igarashi, A.; Ibrahim, O.M.; Inaba, T.; Shimizu, T.; Noda, S.; Obata, H.; et al. Age-related dysfunction of the lacrimal gland and oxidative stress: Evidence from the Cu,Zn-superoxide dismutase-1 (Sod1) knockout mice. Am. J. Pathol. 2012, 180, 1879–1896. [Google Scholar] [CrossRef]
- Uchino, Y.; Kawakita, T.; Miyazawa, M.; Ishii, T.; Onouchi, H.; Yasuda, K.; Ogawa, Y.; Shimmura, S.; Ishii, N.; Tsubota, K. Oxidative stress induced inflammation initiates functional decline of tear production. PLoS ONE 2012, 7, e45805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaser, H.; Dostert, C.; Mak, T.W.; Brenner, D. TNF and ROS Crosstalk in Inflammation. Trends Cell Biol. 2016, 26, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Rocha, E.M.; Alves, M.; Rios, J.D.; Dartt, D.A. The aging lacrimal gland: Changes in structure and function. Ocul. Surf. 2008, 6, 162–174. [Google Scholar] [CrossRef] [Green Version]
- Nichols, K.K.; Foulks, G.N.; Bron, A.J.; Glasgow, B.J.; Dogru, M.; Tsubota, K.; Lemp, M.A.; Sullivan, D.A. The international workshop on meibomian gland dysfunction: Executive summary. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1922–1929. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, O.M.; Dogru, M.; Matsumoto, Y.; Igarashi, A.; Kojima, T.; Wakamatsu, T.H.; Inaba, T.; Shimizu, T.; Shimazaki, J.; Tsubota, K. Oxidative stress induced age dependent meibomian gland dysfunction in Cu, Zn-superoxide dismutase-1 (Sod1) knockout mice. PLoS ONE 2014, 9, e993282014. [Google Scholar] [CrossRef] [Green Version]
- Macri, A.; Scanarotti, C.; Bassi, A.M.; Giuffrida, S.; Sangalli, G.; Traverso, C.E.; Iester, M. Evaluation of oxidative stress levels in the conjunctival epithelium of patients with or without dry eye, and dry eye patients treated with preservative-free hyaluronic acid 0.15 % and vitamin B12 eye drops. Graefe’s Arch. Clin. Exp. Ophthalmol. 2015, 253, 425–430. [Google Scholar] [CrossRef]
- Horwath-Winter, J.; Schmut, O.; Haller-Schober, E.M.; Gruber, A.; Rieger, G. Iodide iontophoresis as a treatment for dry eye syndrome. Br. J. Ophthalmol. 2005, 89, 40–44. [Google Scholar] [CrossRef] [Green Version]
- Downie, L.E.; Ng, S.M.; Lindsley, K.B.; Akpek, E.K. Omega-3 and omega-6 polyunsaturated fatty acids for dry eye disease. Cochrane Database Syst. Rev. 2019, 12, Cd011016. [Google Scholar] [CrossRef]
- Wang, B.; Zuo, X.; Peng, L.; Wang, X.; Zeng, H.; Zhong, J.; Li, S.; Xiao, Y.; Wang, L.; Ouyang, H.; et al. Melatonin ameliorates oxidative stress-mediated injuries through induction of HO-1 and restores autophagic flux in dry eye. Exp. Eye Res. 2021, 205, 108491. [Google Scholar] [CrossRef]
- Sullivan, D.A.; Rocha, E.M.; Aragona, P.; Clayton, J.A.; Ding, J.; Golebiowski, B.; Hampel, U.; McDermott, A.M.; Schaumberg, D.A.; Srinivasan, S.; et al. TFOS DEWS II Sex, Gender, and Hormones Report. Ocul. Surf. 2017, 15, 284–333. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Fjærvoll, H.K.; Fjærvoll, K.A.; Wang, N.H.; Utheim, T.P.; Serhan, C.N.; Dartt, D.A. Sex-based differences in conjunctival goblet cell responses to pro-inflammatory and pro-resolving mediators. Sci. Rep. 2022, 12, 16305. [Google Scholar] [CrossRef]
- Garriz, A.; Morokuma, J.; Bowman, M.; Pagni, S.; Zoukhri, D. Effects of proinflammatory cytokines on lacrimal gland myoepithelial cells contraction. Front. Ophthalmol. 2022, 2, 873486. [Google Scholar] [CrossRef] [PubMed]
- McKay, T.B.; Priyadarsini, S.; Karamichos, D. Sex Hormones, Growth Hormone, and the Cornea. Cells 2022, 11, 224. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, H.; Heydarian, S.; Hooshmand, E.; Saatchi, M.; Yekta, A.; Aghamirsalim, M.; Valadkhan, M.; Mortazavi, M.; Hashemi, A.; Khabazkhoob, M. The prevalence and risk factors for keratoconus: A systematic review and meta-analysis. Cornea 2020, 39, 263–270. [Google Scholar] [CrossRef]
- Tur, V.M.; MacGregor, C.; Jayaswal, R.; O’Brart, D.; Maycock, N. A review of keratoconus: Diagnosis, pathophysiology, and genetics. Surv. Ophthalmol. 2017, 62, 770–783. [Google Scholar]
- De Barros, M.R.M.; Chakravarti, S. Pathogenesis of keratoconus: NRF2-antioxidant, extracellular matrix and cellular dysfunctions. Exp. Eye Res. 2022, 219, 109062. [Google Scholar]
- Navel, V.; Malecaze, J.; Pereira, B.; Baker, J.S.; Malecaze, F.; Sapin, V.; Chiambaretta, F.; Dutheil, F. Oxidative and antioxidative stress markers in keratoconus: A systematic review and meta-analysis. Acta Ophthalmol. 2021, 99, e777–e794. [Google Scholar] [CrossRef]
- Arnal, E.; Peris-Martínez, C.; Menezo, J.L.; Johnsen-Soriano, S.; Romero, F.J. Oxidative stress in keratoconus? Invest. Ophthalmol. Vis. Sci. 2011, 52, 8592–8597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balmus, I.M.; Alexa, A.I.; Ciuntu, R.E.; Danielescu, C.; Stoica, B.; Cojocaru, S.I.; Ciobica, A.; Cantemir, A. Oxidative stress markers dynamics in keratoconus patients’ tears before and after corneal collagen crosslinking procedure. Exp. Eye Res. 2020, 190, 107897. [Google Scholar] [CrossRef] [PubMed]
- López-López, M.; Regueiro, U.; Bravo, S.B.; Chantada-Vázquez, M.D.P.; Varela-Fernández, R.; Ávila-Gómez, P.; Hervella, P.; Lema, I. Tear Proteomics in Keratoconus: A Quantitative SWATH-MS Analysis. Investig. Ophthalmol. Vis. Sci. 2021, 62, 30. [Google Scholar] [CrossRef] [PubMed]
- Toprak, I.; Kucukatay, V.; Yildirim, C.; Kilic-Toprak, E.; Kilic-Erkek, O. Increased systemic oxidative stress in patients with keratoconus. Eye 2014, 28, 285–289. [Google Scholar] [CrossRef] [Green Version]
- Karamichos, D.; Hutcheon, A.E.; Rich, C.B.; Trinkaus-Randall, V.; Asara, J.M.; Zieske, J.D. In vitro model suggests oxidative stress involved in keratoconus disease. Sci. Rep. 2014, 4, 4608. [Google Scholar] [CrossRef] [Green Version]
- Shinde, V.; Hu, N.; Mahale, A.; Maiti, G.; Daoud, Y.; Eberhart, C.G.; Maktabi, A.; Jun, A.S.; Al-Swailem, S.A.; Chakravarti, S. RNA sequencing of corneas from two keratoconus patient groups identifies potential biomarkers and decreased NRF2-antioxidant responses. Sci. Rep. 2020, 10, 9907. [Google Scholar] [CrossRef] [PubMed]
- Lupasco, T.; He, Z.; Cassagne, M.; Sagnial, T.; Brion, L.; Fournié, P.; Gain, P.; Thuret, G.; Allouche, M.; Malecaze, F.; et al. Corneal epithelium in keratoconus underexpresses active NRF2 and a subset of oxidative stress-related genes. PLoS ONE 2022, 17, e0273807. [Google Scholar] [CrossRef]
- Chaerkady, R.; Shao, H.; Scott, S.G.; Pandey, A.; Jun, A.S.; Chakravarti, S. The keratoconus corneal proteome: Loss of epithelial integrity and stromal degeneration. J. Proteom. 2013, 87, 122–131. [Google Scholar] [CrossRef] [Green Version]
- Rabinowitz, Y.S. Keratoconus. Surv. Ophthalmol. 1998, 42, 297–319. [Google Scholar] [CrossRef]
- Wisse, R.P.L.; Kuiper, J.J.W.; Radstake, T.R.D.; Broen, J.C.A. Quantification of Double Stranded DNA Breaks and Telomere Length as Proxies for Corneal Damage and Replicative Stress in Human Keratoconus Corneas. Transl. Vis. Sci. Technol. 2019, 8, 10. [Google Scholar] [CrossRef] [Green Version]
- Kenney, M.C.; Brown, D.J.; Rajeev, B. Everett Kinsey lecture. The elusive causes of keratoconus: A working hypothesis. Clao J. 2000, 26, 10–13. [Google Scholar] [PubMed]
- Behndig, A.; Karlsson, K.; Johansson, B.O.; Brännström, T.; Marklund, S.L. Superoxide dismutase isoenzymes in the normal and diseased human cornea. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2293–2296. [Google Scholar] [PubMed]
- Udar, N.; Atilano, S.R.; Small, K.; Nesburn, A.B.; Kenney, M.C. SOD1 haplotypes in familial keratoconus. Cornea 2009, 28, 902–907. [Google Scholar] [CrossRef] [PubMed]
- Udar, N.; Atilano, S.R.; Brown, D.J.; Holguin, B.; Small, K.; Nesburn, A.B.; Kenney, M.C. SOD1: A candidate gene for keratoconus. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3345–3351. [Google Scholar] [CrossRef] [Green Version]
- Moschos, M.M.; Kokolakis, N.; Gazouli, M.; Chatziralli, I.P.; Droutsas, D.; Anagnou, N.P.; Ladas, I.D. Polymorphism Analysis of VSX1 and SOD1 Genes in Greek Patients with Keratoconus. Ophthalmic Genet. 2015, 36, 213–217. [Google Scholar] [CrossRef]
- Abu-Amero, K.K.; Azad, T.A.; Kalantan, H.; Sultan, T.; Al-Muammar, A.M. Mitochondrial sequence changes in keratoconus patients. Investig. Ophthalmol. Vis. Sci. 2014, 55, 1706–1710. [Google Scholar] [CrossRef] [Green Version]
- Atilano, S.R.; Coskun, P.; Chwa, M.; Jordan, N.; Reddy, V.; Le, K.; Wallace, D.C.; Kenney, M.C. Accumulation of mitochondrial DNA damage in keratoconus corneas. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1256–1263. [Google Scholar] [CrossRef] [Green Version]
- Gadelha, D.N.B.; Feitosa, A.F.B.; da Silva, R.G.; Antunes, L.T.; Muniz, M.C.; de Oliveira, M.A.; Andrade, D.O.; da Paz Silva, N.M.; Cronemberger, S.; Schamber-Reis, B.L.F. Screening for Novel LOX and SOD1 Variants in Keratoconus Patients from Brazil. J. Ophthalmic Vis. Res. 2020, 15, 138–148. [Google Scholar] [CrossRef]
- Nejabat, M.; Naghash, P.; Dastsooz, H.; Mohammadi, S.; Alipour, M.; Fardaei, M. VSX1 and SOD1 Mutation Screening in Patients with Keratoconus in the South of Iran. J. Ophthalmic Vis. Res. 2017, 12, 135–140. [Google Scholar] [CrossRef]
- Stabuc-Silih, M.; Strazisar, M.; Hawlina, M.; Glavac, D. Absence of pathogenic mutations in VSX1 and SOD1 genes in patients with keratoconus. Cornea 2010, 29, 172–176. [Google Scholar] [CrossRef]
- Al-Muammar, A.M.; Kalantan, H.; Azad, T.A.; Sultan, T.; Abu-Amero, K.K. Analysis of the SOD1 gene in keratoconus patients from Saudi Arabia. Ophthalmic Genet. 2015, 36, 373–375. [Google Scholar] [CrossRef] [PubMed]
- Bykhovskaya, Y.; Rabinowitz, Y.S. Update on the genetics of keratoconus. Exp. Eye Res. 2021, 202, 108398. [Google Scholar] [CrossRef] [PubMed]
- Lema, I.; Durán, J.A. Inflammatory molecules in the tears of patients with keratoconus. Ophthalmology 2005, 112, 654–659. [Google Scholar] [CrossRef]
- Daphne Teh, A.L.; Jayapalan, J.J.; Loke, M.F.; Wan Abdul Kadir, A.J.; Subrayan, V. Identification of potential serum metabolic biomarkers for patient with keratoconus using untargeted metabolomics approach. Exp. Eye Res. 2021, 211, 108734. [Google Scholar] [CrossRef] [PubMed]
- McKay, T.B.; Hjortdal, J.; Sejersen, H.; Asara, J.M.; Wu, J.; Karamichos, D. Endocrine and Metabolic Pathways Linked to Keratoconus: Implications for the Role of Hormones in the Stromal Microenvironment. Sci. Rep. 2016, 6, 25534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamali, H.; Heydari, M.; Masihpour, N.; Khosravi, A.; Zare, M.; Shams, M.; Omrani, G.R. Serum androgens and prolactin levels in patients with keratoconus. Clin. Exp. Optom. 2022, 1–5. [Google Scholar] [CrossRef]
- Ayan, B.; Yuksel, N.; Carhan, A.; Gumuşkaya Ocal, B.; Akcay, E.; Cagil, N.; Asik, M.D. Evaluation estrogen, progesteron and androgen receptor expressions in corneal epithelium in keratoconus. Contact Lens Anterior Eye 2019, 42, 492–496. [Google Scholar] [CrossRef]
- Karamichos, D.; Escandon, P.; Vasini, B.; Nicholas, S.E.; Van, L.; Dang, D.H.; Cunningham, R.L.; Riaz, K.M. Anterior pituitary, sex hormones, and keratoconus: Beyond traditional targets. Prog. Retin. Eye Res. 2022, 88, 101016. [Google Scholar] [CrossRef]
- Lasagni Vitar, R.M.; Bonelli, F.; Rama, P.; Ferrari, G. Nutritional and Metabolic Imbalance in Keratoconus. Nutrients 2022, 14, 913. [Google Scholar] [CrossRef]
- Karamichos, D.; Zareian, R.; Guo, X.; Hutcheon, A.E.; Ruberti, J.W.; Zieske, J.D. Novel in Vitro Model for Keratoconus Disease. J. Funct. Biomater. 2012, 3, 760–775. [Google Scholar] [CrossRef] [Green Version]
- Atilano, S.R.; Lee, D.H.; Fukuhara, P.S.; Chwa, M.; Nesburn, A.B.; Udar, N.; Kenney, M.C. Corneal Oxidative Damage in Keratoconus Cells due to Decreased Oxidant Elimination from Modified Expression Levels of SOD Enzymes, PRDX6, SCARA3, CPSF3, and FOXM1. J. Ophthalmic Vis. Res. 2019, 14, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ismail, S.; McGhee, J.J.; Wadhwa, H.; Noord, N.; van der Werf, B.; Sherwin, T. Differences in sphere-forming cells from keratoconic and normal corneal tissue: Implications for keratoconus pathogenesis. Exp. Eye Res. 2021, 202, 108301. [Google Scholar] [CrossRef]
- Morishige, N.; Magome, K.; Ueno, A.; Matsui, T.A.; Nishida, T. Relations Among Corneal Curvature, Thickness, and Volume in Keratoconus as Evaluated by Anterior Segment-Optical Coherence Tomography. Investig. Ophthalmol. Vis. Sci. 2019, 60, 3794–3802. [Google Scholar] [CrossRef] [Green Version]
- Wojakowska, A.; Pietrowska, M.; Widlak, P.; Dobrowolski, D.; Wylęgała, E.; Tarnawska, D. Metabolomic Signature Discriminates Normal Human Cornea from Keratoconus-A Pilot GC/MS Study. Molecules 2020, 25, 2933. [Google Scholar] [CrossRef]
- McKay, T.B.; Sarker-Nag, A.; Lyon, D.; Asara, J.M.; Karamichos, D. Quercetin modulates keratoconus metabolism in vitro. Cell Biochem. Funct. 2015, 33, 341–350. [Google Scholar] [CrossRef] [Green Version]
- McKay, T.B.; Lyon, D.; Sarker-Nag, A.; Priyadarsini, S.; Asara, J.M.; Karamichos, D. Quercetin attenuates lactate production and extracellular matrix secretion in keratoconus. Sci. Rep. 2015, 5, 9003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKay, T.B.; Kivanany, P.B.; Nicholas, S.E.; Nag, O.K.; Elliott, M.H.; Petroll, W.M.; Karamichos, D. Quercetin Decreases Corneal Haze In Vivo and Influences Gene Expression of TGF-Beta Mediators In Vitro. Metabolites 2022, 12, 3673. [Google Scholar] [CrossRef]
- Sağlik, A.; Koyuncu, İ.; Soydan, A.; Sağlik, F.; Gönel, A. Tear Organic Acid Analysis After Corneal Collagen Crosslinking in Keratoconus. Eye Contact Lens 2020, 46 (Suppl. S2), S122–S128. [Google Scholar] [CrossRef] [PubMed]
- Sharif, R.; Sejersen, H.; Frank, G.; Hjortdal, J.; Karamichos, D. Effects of collagen cross-linking on the keratoconus metabolic network. Eye 2018, 32, 1271–1281. [Google Scholar] [CrossRef] [Green Version]
- Stachon, T.; Kolev, K.; Flaskó, Z.; Seitz, B.; Langenbucher, A.; Szentmáry, N. Arginase activity, urea, and hydroxyproline concentration are reduced in keratoconus keratocytes. Graefe’s Arch. Clin. Exp. Ophthalmol. 2017, 255, 91–97. [Google Scholar] [CrossRef]
- Foster, J.W.; Shinde, V.; Soiberman, U.S.; Sathe, G.; Liu, S.; Wan, J.; Qian, J.; Dauoud, Y.; Pandey, A.; Jun, A.S.; et al. Integrated Stress Response and Decreased ECM in Cultured Stromal Cells From Keratoconus Corneas. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2977–2986. [Google Scholar] [CrossRef] [Green Version]
- McKay, T.B.; Priyadarsini, S.; Rowsey, T.; Karamichos, D. Arginine Supplementation Promotes Extracellular Matrix and Metabolic Changes in Keratoconus. Cells 2021, 10, 2076. [Google Scholar] [CrossRef]
- Wojcik, K.A.; Kaminska, A.; Blasiak, J.; Szaflik, J.; Szaflik, J.P. Oxidative stress in the pathogenesis of keratoconus and Fuchs endothelial corneal dystrophy. Int. J. Mol. Sci. 2013, 14, 19294–19308. [Google Scholar] [CrossRef] [Green Version]
- Buddi, R.; Lin, B.; Atilano, S.R.; Zorapapel, N.C.; Kenney, M.C.; Brown, D.J. Evidence of oxidative stress in human corneal diseases. J. Histochem. Cytochem. 2002, 50, 341–351. [Google Scholar] [CrossRef]
- Ong Tone, S.; Kocaba, V.; Böhm, M.; Wylegala, A.; White, T.L.; Jurkunas, U.V. Fuchs endothelial corneal dystrophy: The vicious cycle of Fuchs pathogenesis. Prog. Retin. Eye Res. 2021, 80, 100863. [Google Scholar] [CrossRef]
- Lovicu, F.J.; McAvoy, J.W. Growth factor regulation of lens development. Dev. Biol. 2005, 280, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Foster, A.; Resnikoff, S. The impact of Vision 2020 on global blindness. Eye 2005, 19, 1133–1135. [Google Scholar] [CrossRef] [Green Version]
- Gupta, V.B.; Rajagopala, M.; Ravishankar, B. Etiopathogenesis of cataract: An appraisal. Indian J. Ophthalmol. 2014, 62, 103–110. [Google Scholar] [CrossRef]
- Wormstone, I.M. Posterior capsule opacification: A cell biological perspective. Exp. Eye Res. 2002, 74, 337–347. [Google Scholar] [CrossRef]
- Wride, M.A. Lens fibre cell differentiation and organelle loss: Many paths lead to clarity. Philos. Trans. R Soc. Lond. B Biol. Sci. 2011, 366, 1219–1233. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Edassery, S.L.; Ali, L.; Thomson, B.R.; Savas, J.N.; Jin, J. Long-lived metabolic enzymes in the crystalline lens identified by pulse-labeling of mice and mass spectrometry. eLife 2019, 8, e50170. [Google Scholar] [CrossRef] [PubMed]
- McNulty, R.; Wang, H.; Mathias, R.T.; Ortwerth, B.J.; Truscott, R.J.; Bassnett, S. Regulation of tissue oxygen levels in the mammalian lens. J. Physiol. 2004, 559, 883–898. [Google Scholar] [CrossRef]
- Lim, J.C.; Grey, A.C.; Zahraei, A.; Donaldson, P.J. Age-dependent changes in glutathione metabolism pathways in the lens: New insights into therapeutic strategies to prevent cataract formation-A review. Clin. Exp. Ophthalmol. 2020, 48, 1031–1042. [Google Scholar] [CrossRef]
- Zhao, H.; Brown, P.H.; Magone, M.T.; Schuck, P. The molecular refractive function of lens γ-crystallins. J. Mol. Biol. 2011, 411, 680–699. [Google Scholar] [CrossRef] [Green Version]
- Mills-Henry, I.A.; Thol, S.L.; Kosinski-Collins, M.S.; Serebryany, E.; King, J.A. Kinetic stability of long-lived human lens γ-crystallins and their isolated double Greek key domains. Biophys. J. 2019, 117, 269–280. [Google Scholar] [CrossRef]
- Truscott, R.J.W.; Friedrich, M.G. Molecular Processes Implicated in Human Age-Related Nuclear Cataract. Investig. Ophthalmol. Vis. Sci. 2019, 60, 5007–5021. [Google Scholar] [CrossRef] [Green Version]
- Schey, K.L.; Wang, Z.; Friedrich, M.G.; Truscott, R.J.W. New insights into the mechanisms of age-related protein-protein crosslinking in the human lens. Exp. Eye Res. 2021, 209, 108679. [Google Scholar] [CrossRef]
- Gakamsky, A.; Duncan, R.R.; Howarth, N.M.; Dhillon, B.; Buttenschön, K.K.; Daly, D.J.; Gakamsky, D. Tryptophan and Non-Tryptophan Fluorescence of the Eye Lens Proteins Provides Diagnostics of Cataract at the Molecular Level. Sci. Rep. 2017, 7, 40375. [Google Scholar] [CrossRef] [Green Version]
- Modenese, A.; Gobba, F. Cataract frequency and subtypes involved in workers assessed for their solar radiation exposure: A systematic review. Acta Ophthalmol. 2018, 96, 779–788. [Google Scholar] [CrossRef] [Green Version]
- Cekic, O. Effect of cigarette smoking on copper, lead, and cadmium accumulation in human lens. Br. J. Ophthalmol. 1998, 82, 186–188. [Google Scholar] [CrossRef] [Green Version]
- Srikanthan, D.; Bateman, O.A.; Purkiss, A.G.; Slingsby, C. Sulfur in human crystallins. Exp. Eye Res. 2004, 79, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Truscott, R.J. Age-related nuclear cataract-oxidation is the key. Exp. Eye Res. 2005, 80, 709–725. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.J. Disulphide cross-linked protein of high molecular weight in human cataractous lens. Exp. Eye Res. 1973, 17, 377–383. [Google Scholar] [CrossRef]
- Spector, A.; Roy, D. Disulfide-linked high molecular weight protein associated with human cataract. Proc. Natl. Acad. Sci. USA 1978, 75, 3244–3248. [Google Scholar] [CrossRef] [Green Version]
- Truscott, R.J.; Augusteyn, R.C. Oxidative changes in human lens proteins during senile nuclear cataract formation. Biochim. Biophys. Acta 1977, 492, 43–52. [Google Scholar] [CrossRef]
- Hains, P.G.; Truscott, R.J. Post-translational modifications in the nuclear region of young, aged, and cataract human lenses. J. Proteome Res. 2007, 6, 3935–3943. [Google Scholar] [CrossRef]
- Lampi, K.J.; Ma, Z.; Hanson, S.R.; Azuma, M.; Shih, M.; Shearer, T.R.; Smith, D.L.; Smith, J.B.; David, L.L. Age-related changes in human lens crystallins identified by two-dimensional electrophoresis and mass spectrometry. Exp. Eye Res. 1998, 67, 31–43. [Google Scholar] [CrossRef]
- Norton-Baker, B.; Mehrabi, P.; Kwok, A.O.; Roskamp, K.W.; Rocha, M.A.; Sprague-Piercy, M.A.; von Stetten, D.; Miller, R.J.D.; Martin, R.W. Deamidation of the human eye lens protein gammaS-crystallin accelerates oxidative aging. Structure 2022, 30, 763–776.e4. [Google Scholar] [CrossRef]
- Vetter, C.J.; Thorn, D.C.; Wheeler, S.G.; Mundorff, C.; Halverson, K.; Wales, T.E.; Shinde, U.; Engen, J.R.; David, L.L.; Carver, J.A.; et al. Cumulative deamidations of the major lens protein γS-crystallin increase its aggregation during unfolding and oxidation. Protein Sci. 2020, 29, 1945–1963. [Google Scholar] [CrossRef]
- Zhao, W.J.; Yan, Y.B. Increasing susceptibility to oxidative stress by cataract-causing crystallin mutations. Int. J. Biol. Macromol. 2018, 108, 665–673. [Google Scholar] [CrossRef]
- Serebryany, E.; Woodard, J.C.; Adkar, B.V.; Shabab, M.; King, J.A.; Shakhnovich, E.I. An internal disulfide locks a misfolded aggregation-prone intermediate in cataract-linked mutants of human γD-crystallin. J. Biol. Chem. 2016, 291, 19172–19183. [Google Scholar] [CrossRef] [Green Version]
- Serebryany, E.; Thorn, D.C.; Quintanar, L. Redox chemistry of lens crystallins: A system of cysteines. Exp. Eye Res. 2021, 211, 108707. [Google Scholar] [CrossRef] [PubMed]
- Michael, R.; Bron, A.J. The ageing lens and cataract: A model of normal and pathological ageing. Philos. Trans. R Soc. Lond. B Biol. Sci. 2011, 366, 1278–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giblin, F.J. Glutathione: A vital lens antioxidant. J. Ocul. Pharmacol. Ther. 2000, 16, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Lou, M.F. Redox regulation in the lens. Prog. Retin. Eye Res. 2003, 22, 657–682. [Google Scholar] [CrossRef]
- Reddan, J.R.; Giblin, F.J.; Kadry, R.; Leverenz, V.R.; Pena, J.T.; Dziedzic, D.C. Protection from oxidative insult in glutathione depleted lens epithelial cells. Exp. Eye Res. 1999, 68, 117–127. [Google Scholar] [CrossRef]
- Sweeney, M.H.; Truscott, R.J. An impediment to glutathione diffusion in older normal human lenses: A possible precondition for nuclear cataract. Exp. Eye Res. 1998, 67, 587–595. [Google Scholar] [CrossRef]
- Fan, X.; Liu, X.; Hao, S.; Wang, B.; Robinson, M.L.; Monnier, V.M. The LEGSKO mouse: A mouse model of age-related nuclear cataract based on genetic suppression of lens glutathione synthesis. PLoS ONE 2012, 7, e50832. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Zhou, S.; Wang, B.; Hom, G.; Guo, M.; Li, B.; Yang, J.; Vaysburg, D.; Monnier, V.M. Evidence of highly conserved β-crystallin disulfidome that can be mimicked by in vitro oxidation in age-related human cataract and glutathione depleted mouse lens. Mol. Cell. Proteom. 2015, 14, 3211–3223. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Hom, G.; Zhou, S.; Guo, M.; Li, B.; Yang, J.; Monnier, V.M.; Fan, X. The oxidized thiol proteome in aging and cataractous mouse and human lens revealed by ICAT labeling. Aging Cell 2017, 16, 244–261. [Google Scholar] [CrossRef]
- Roskamp, K.W.; Azim, S.; Kassier, G.; Norton-Baker, B.; Sprague-Piercy, M.A.; Miller, R.J.D.; Martin, R.W. Human γS-crystallin-copper binding helps buffer against aggregation caused by oxidative damage. Biochemistry 2020, 59, 2371–2385. [Google Scholar] [CrossRef] [PubMed]
- Serebryany, E.; Yu, S.; Trauger, S.A.; Budnik, B.; Shakhnovich, E.I. Dynamic disulfide exchange in a crystallin protein in the human eye lens promotes cataract-associated aggregation. J. Biol. Chem. 2018, 293, 17997–18009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skouri-Panet, F.; Bonnete, F.; Prat, K.; Bateman, O.A.; Lubsen, N.H.; Tardieu, A. Lens crystallins and oxidation: The special case of γS. Biophys. Chem. 2001, 89, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Thorn, D.C.; Grosas, A.B.; Mabbitt, P.D.; Ray, N.J.; Jackson, C.J.; Carver, J.A. The structure and stability of the disulfide-linked γS-crystallin dimer provide insight into oxidation products associated with lens cataract formation. J. Mol. Biol. 2019, 431, 483–497. [Google Scholar] [CrossRef]
- Quinlan, R.A.; Hogg, P.J. γ-Crystallin redox-detox in the lens. J. Biol. Chem. 2018, 293, 18010–18011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelkader, H.; Alany, R.G.; Pierscionek, B. Age-related cataract and drug therapy: Opportunities and challenges for topical antioxidant delivery to the lens. J. Pharm. Pharmacol. 2015, 67, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Serebryany, E.; Chowdhury, S.; Woods, C.N.; Thorn, D.C.; Watson, N.E.; McClelland, A.A.; Klevit, R.E.; Shakhnovich, E.I. A native chemical chaperone in the human eye lens. eLife 2022, 11, e76923. [Google Scholar] [CrossRef]
- Fan, X.; Monnier, V.M.; Whitson, J. Lens glutathione homeostasis: Discrepancies and gaps in knowledge standing in the way of novel therapeutic approaches. Exp. Eye Res. 2017, 156, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Shu, D.Y.; Ong, K.; Lovicu, F.J. Histopathology of Subcapsular Cataract in a Patient with Atopic Dermatitis. Optom. Vis. Sci. 2017, 94, 270–276. [Google Scholar] [CrossRef]
- Shu, D.Y.; Lovicu, F.J. Enhanced EGF receptor-signaling potentiates TGFβ-induced lens epithelial-mesenchymal transition. Exp. Eye Res. 2019, 185, 107693. [Google Scholar] [CrossRef]
- Shu, D.Y.; Wojciechowski, M.; Lovicu, F.J. ERK1/2-mediated EGFR-signaling is required for TGFβ-induced lens epithelial-mesenchymal transition. Exp. Eye Res. 2019, 178, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Shu, D.Y.; Wojciechowski, M.C.; Lovicu, F.J. Bone Morphogenetic Protein-7 Suppresses TGFβ2-Induced Epithelial-Mesenchymal Transition in the Lens: Implications for Cataract Prevention. Investig. Ophthalmol. Vis. Sci. 2017, 58, 781–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojciechowski, M.C.; Mahmutovic, L.; Shu, D.Y.; Lovicu, F.J. ERK1/2 signaling is required for the initiation but not progression of TGFβ-induced lens epithelial to mesenchymal transition (EMT). Exp. Eye Res. 2017, 159, 98–113. [Google Scholar] [CrossRef]
- Shu, D.Y.; Lovicu, F.J. Insights into Bone Morphogenetic Protein-(BMP-) Signaling in Ocular Lens Biology and Pathology. Cells 2021, 10, 2604. [Google Scholar] [CrossRef] [PubMed]
- Shu, D.Y.; Ng, K.; Wishart, T.F.L.; Chui, J.; Lundmark, M.; Flokis, M.; Lovicu, F.J. Contrasting roles for BMP-4 and ventromorphins (BMP agonists) in TGFbeta-induced lens EMT. Exp. Eye Res. 2021, 206, 108546. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yan, H.; Chen, Y.; Li, G.; Bin, Y.; Zhou, X. Moderate oxidative stress promotes epithelial-mesenchymal transition in the lens epithelial cells via the TGF-beta/Smad and Wnt/beta-catenin pathways. Mol. Cell. Biochem. 2021, 476, 1631–1642. [Google Scholar] [CrossRef]
- Chamberlain, C.G.; Mansfield, K.J.; Cerra, A. Glutathione and catalase suppress TGFbeta-induced cataract-related changes in cultured rat lenses and lens epithelial explants. Mol. Vis. 2009, 15, 895–905. [Google Scholar]
- Wei, Z.; Caty, J.; Whitson, J.; Zhang, A.D.; Srinivasagan, R.; Kavanagh, T.J.; Yan, H.; Fan, X. Reduced Glutathione Level Promotes Epithelial-Mesenchymal Transition in Lens Epithelial Cells via a Wnt/beta-Catenin-Mediated Pathway: Relevance for Cataract Therapy. Am. J. Pathol. 2017, 187, 2399–2412. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Li, J.; Zhang, X.; Zhang, X.; Zhang, X.; Zhu, Y.; Chen, C.; Liu, Z.; Wu, X.; Wang, D.; et al. Extracellular vesicles promote epithelial-to-mesenchymal transition of lens epithelial cells under oxidative stress. Exp. Cell Res. 2021, 398, 112362. [Google Scholar] [CrossRef]
- Thompson, B.; Davidson, E.A.; Chen, Y.; Orlicky, D.J.; Thompson, D.C.; Vasiliou, V. Oxidative stress induces inflammation of lens cells and triggers immune surveillance of ocular tissues. Chem. Biol. Interact. 2022, 355, 109804. [Google Scholar] [CrossRef]
- Das, S.J.; Lovicu, F.J.; Collinson, E.J. Nox4 Plays a Role in TGF-beta-Dependent Lens Epithelial to Mesenchymal Transition. Investig. Ophthalmol. Vis. Sci. 2016, 57, 3665–3673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Wikstrom, P.; Walum, E.; Lovicu, F.J. A novel NADPH oxidase inhibitor targeting Nox4 in TGFbeta-induced lens epithelial to mesenchymal transition. Exp. Eye Res. 2019, 185, 107692. [Google Scholar] [CrossRef] [PubMed]
- McCaa, C.S. The eye and visual nervous system: Anatomy, physiology and toxicology. Environ. Health Perspect. 1982, 44, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Masland, R.H. The fundamental plan of the retina. Nat. Neurosci. 2001, 4, 877–886. [Google Scholar] [CrossRef]
- Gollisch, T.; Meister, M. Eye smarter than scientists believed: Neural computations in circuits of the retina. Neuron 2010, 65, 150–164. [Google Scholar] [CrossRef] [Green Version]
- Masland, R.H. The neuronal organization of the retina. Neuron 2012, 76, 266–280. [Google Scholar] [CrossRef] [Green Version]
- Molday, R.S.; Moritz, O.L. Photoreceptors at a glance. J. Cell Sci. 2015, 128, 4039–4045. [Google Scholar] [CrossRef] [Green Version]
- Demb, J.B.; Singer, J.H. Functional Circuitry of the Retina. Annu. Rev. Vis. Sci. 2015, 1, 263–289. [Google Scholar] [CrossRef] [Green Version]
- Flores-Herr, N.; Protti, D.A.; Wässle, H. Synaptic currents generating the inhibitory surround of ganglion cells in the mammalian retina. J. Neurosci. 2001, 21, 4852–4863. [Google Scholar] [CrossRef] [Green Version]
- Bruesch, S.R.; Arey, L.B. The number of myelinated and unmyelinated fibres in the optic nerve of vertebrates. J. Comp. Neurol. 1943, 77, 631. [Google Scholar] [CrossRef]
- Garron, L.K. The ultrastructure of the retinal pigment epithelium with observations on the choriocapillaris and Bruch’s membrane. Trans. Am. Ophthalmol. Soc. 1963, 61, 545. [Google Scholar]
- Gu, X.; Neric, N.J.; Crabb, J.S.; Crabb, J.W.; Bhattacharya, S.K.; Rayborn, M.E.; Hollyfield, J.G.; Bonilha, V.L. Age-related changes in the retinal pigment epithelium (RPE). PLoS ONE 2012, 7, e38673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kur, J.; Newman, E.A.; Chan-Ling, T. Cellular and physiological mechanisms underlying blood flow regulation in the retina and choroid in health and disease. Prog. Retin. Eye Res. 2012, 31, 377–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Yang, P.; Kijlstra, A. Distribution, markers, and functions of retinal microglia. Ocul. Immunol. Inflamm. 2002, 10, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Reichenbach, A.; Bringmann, A. Role of Purines in Müller Glia. J. Ocul. Pharmacol. Ther. 2016, 32, 518–533. [Google Scholar] [CrossRef]
- Rathnasamy, G.; Foulds, W.S.; Ling, E.-A.; Kaur, C. Retinal microglia—A key player in healthy and diseased retina. Prog. Neurobiol. 2019, 173, 18–40. [Google Scholar] [CrossRef]
- Reichenbach, A.; Bringmann, A. Glia of the human retina. Glia 2020, 68, 768–796. [Google Scholar] [CrossRef]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [Green Version]
- Provis, J.M. Development of the Primate Retinal Vasculature. Prog. Retin. Eye Res. 2001, 20, 799–821. [Google Scholar] [CrossRef]
- Bessis, A.; Béchade, C.; Bernard, D.; Roumier, A. Microglial control of neuronal death and synaptic properties. Glia 2007, 55, 233–238. [Google Scholar] [CrossRef]
- Langmann, T. Microglia activation in retinal degeneration. J. Leukoc. Biol. 2007, 81, 1345–1351. [Google Scholar] [CrossRef] [PubMed]
- Büssow, H. The astrocytes in the retina and optic nerve head of mammals: A special glia for the ganglion cell axons. Cell Tissue Res. 1980, 206, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Distler, C.; Weigel, H.; Hoffmann, K.P. Glia cells of the monkey retina. I. Astrocytes. J. Comp. Neurol. 1993, 333, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Holländer, H.; Makarov, F.; Dreher, Z.; van Driel, D.; Chan-Ling, T.; Stone, J. Structure of the macroglia of the retina: Sharing and division of labour between astrocytes and Müller cells. J. Comp. Neurol. 1991, 313, 587–603. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, H.; Seo, M.S.; Ozaki, K.; Yamada, H.; Yamada, E.; Okamoto, N.; Hofmann, F.; Wood, J.M.; Campochiaro, P.A. Blockade of vascular endothelial cell growth factor receptor signaling is sufficient to completely prevent retinal neovascularization. Am. J. Pathol. 2000, 156, 697–707. [Google Scholar] [CrossRef] [Green Version]
- Stone, J.; Itin, A.; Alon, T.; Pe’er, J.; Gnessin, H.; Chan-Ling, T.; Keshet, E. Development of retinal vasculature is mediated by hypoxia-induced vascular endothelial growth factor (VEGF) expression by neuroglia. J. Neurosci. 1995, 15, 4738–4747. [Google Scholar] [CrossRef]
- Newman, E.; Reichenbach, A. The Müller cell: A functional element of the retina. Trends Neurosci. 1996, 19, 307–312. [Google Scholar] [CrossRef]
- Agte, S.; Junek, S.; Matthias, S.; Ulbricht, E.; Erdmann, I.; Wurm, A.; Schild, D.; Käs, J.A.; Reichenbach, A. Müller glial cell-provided cellular light guidance through the vital guinea-pig retina. Biophys. J. 2011, 101, 2611–2619. [Google Scholar] [CrossRef] [Green Version]
- Reichenbach, A.; Bringmann, A. New functions of Müller cells. Glia 2013, 61, 651–678. [Google Scholar] [CrossRef]
- Huster, D.; Reichenbach, A.; Reichelt, W. The glutathione content of retinal Müller (glial) cells: Effect of pathological conditions. Neurochem. Int. 2000, 36, 461–469. [Google Scholar] [CrossRef]
- Coughlin, B.A.; Feenstra, D.J.; Mohr, S. Müller cells and diabetic retinopathy. Vis. Res. 2017, 139, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Capozzi, M.E.; McCollum, G.W.; Cousins, D.B.; Penn, J.S. Linoleic Acid is a Diabetes-relevant Stimulator of Retinal Inflammation in Human Retinal Muller Cells and Microvascular Endothelial Cells. J. Diabetes Metab. 2016, 7, 718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, D.Y.; Cringle, S.J.; Yu, P.K.; Balaratnasingam, C.; Mehnert, A.; Sarunic, M.V.; An, D.; Su, E.N. Retinal capillary perfusion: Spatial and temporal heterogeneity. Prog. Retin. Eye Res. 2019, 70, 23–54. [Google Scholar] [CrossRef] [PubMed]
- Archer, D.B.; Gardiner, T.A.; Stitt, A.W. Functional anatomy, fine structure and basic pathology of the retinal vasculature. In Retinal Vascular Disease; Springer: Berlin/Heidelberg, Germany, 2007; pp. 3–23. [Google Scholar]
- Giannaccini, M.; Usai, A.; Chiellini, F.; Guadagni, V.; Andreazzoli, M.; Ori, M.; Pasqualetti, M.; Dente, L.; Raffa, V. Neurotrophin-conjugated nanoparticles prevent retina damage induced by oxidative stress. Cell. Mol. Life Sci. 2018, 75, 1255–1267. [Google Scholar] [CrossRef] [Green Version]
- Caprara, C.; Thiersch, M.; Lange, C.; Joly, S.; Samardzija, M.; Grimm, C. HIF1A is essential for the development of the intermediate plexus of the retinal vasculature. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2109–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, J.; Zhang, M.; Hwang, T.; Bailey, S.; Wilson, D.; Jia, Y.; Huang, D. Detailed vascular anatomy of the human retina by projection-resolved optical coherence tomography angiography. Sci. Rep. 2017, 7, 42201. [Google Scholar] [CrossRef] [Green Version]
- Moran, E.P.; Wang, Z.; Chen, J.; Sapieha, P.; Smith, L.E.; Ma, J.-X. Neurovascular cross talk in diabetic retinopathy: Pathophysiological roles and therapeutic implications. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H738–H749. [Google Scholar] [CrossRef] [Green Version]
- Vecino, E.; Rodriguez, F.D.; Ruzafa, N.; Pereiro, X.; Sharma, S.C. Glia–neuron interactions in the mammalian retina. Prog. Retin. Eye Res. 2016, 51, 1–40. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.M.; Maple, B.R. Amino acid neurotransmitters in the retina: A functional overview. Vis. Res. 1998, 38, 1371–1384. [Google Scholar] [CrossRef] [Green Version]
- Selvam, S.; Kumar, T.; Fruttiger, M. Retinal vasculature development in health and disease. Prog. Retin. Eye Res. 2018, 63, 1–19. [Google Scholar] [CrossRef]
- Ames III, A. Energy requirements of CNS cells as related to their function and to their vulnerability to ischemia: A commentary based on studies on retina. Can. J. Physiol. Pharmacol. 1992, 70, S158–S164. [Google Scholar] [CrossRef] [PubMed]
- Maenhaut, N.; Boussery, K.; Delaey, C.; Van de Voorde, J. Control of retinal arterial tone by a paracrine retinal relaxing factor. Microcirculation 2007, 14, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, E.L.; Downie, L.E.; Ly, A.; Ward, M.M.; Batcha, A.H.; Puthussery, T.; Yee, P.; Hatzopoulos, K.M. A review of the role of glial cells in understanding retinal disease. Clin. Exp. Optom. 2008, 91, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Münzel, T.; Camici, G.G.; Maack, C.; Bonetti, N.R.; Fuster, V.; Kovacic, J.C. Impact of Oxidative Stress on the Heart and Vasculature: Part 2 of a 3-Part Series. J. Am. Coll. Cardiol. 2017, 70, 212–229. [Google Scholar] [CrossRef] [PubMed]
- Zadeh, J.K.; Ruemmler, R.; Hartmann, E.K.; Ziebart, A.; Ludwig, M.; Patzak, A.; Xia, N.; Li, H.; Pfeiffer, N.; Gericke, A. Responses of retinal arterioles and ciliary arteries in pigs with acute respiratory distress syndrome (ARDS). Exp. Eye Res. 2019, 184, 152–161. [Google Scholar] [CrossRef]
- Endemann, D.H.; Schiffrin, E.L. Endothelial dysfunction. J. Am. Soc. Nephrol. 2004, 15, 1983–1992. [Google Scholar] [CrossRef] [Green Version]
- Meza, C.A.; La Favor, J.D.; Kim, D.H.; Hickner, R.C. Endothelial Dysfunction: Is There a Hyperglycemia-Induced Imbalance of NOX and NOS? Int. J. Mol. Sci. 2019, 20, 3775. [Google Scholar] [CrossRef] [Green Version]
- Bellezza, I. Oxidative stress in age-related macular degeneration: Nrf2 as therapeutic target. Front. Pharmacol. 2018, 9, 1280. [Google Scholar] [CrossRef]
- Deng, Y.; Qiao, L.; Du, M.; Qu, C.; Wan, L.; Li, J.; Huang, L. Age-related macular degeneration: Epidemiology, genetics, pathophysiology, diagnosis, and targeted therapy. Genes Dis. 2022, 9, 62–79. [Google Scholar] [CrossRef]
- Ehrlich, R.; Harris, A.; Kheradiya, N.S.; Winston, D.M.; Ciulla, T.A.; Wirostko, B. Age-related macular degeneration and the aging eye. Clin. Interv. Aging 2008, 3, 473. [Google Scholar]
- Jager, R.D.; Mieler, W.F.; Miller, J.W. Age-related macular degeneration. N. Engl. J. Med. 2008, 358, 2606–2617. [Google Scholar] [CrossRef] [Green Version]
- Rickman, C.B.; Farsiu, S.; Toth, C.A.; Klingeborn, M. Dry age-related macular degeneration: Mechanisms, therapeutic targets, and imaging. Investig. Ophthalmol. Vis. Sci. 2013, 54, ORSF68–ORSF80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobbs, S.D.; Pierce, K. Wet age-related macular degeneration (Wet AMD). In StatPearls; StatPearls Publishing: Tampa, FL, USA, 2022. [Google Scholar]
- Young, R.W. Pathophysiology of age-related macular degeneration. Surv. Ophthalmol. 1987, 31, 291–306. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.-Y.; Cringle, S.J. Retinal degeneration and local oxygen metabolism. Exp. Eye Res. 2005, 80, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Beatty, S.; Koh, H.-H.; Phil, M.; Henson, D.; Boulton, M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv. Ophthalmol. 2000, 45, 115–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abokyi, S.; To, C.-H.; Lam, T.T.; Tse, D.Y. Central role of oxidative stress in age-related macular degeneration: Evidence from a review of the molecular mechanisms and animal models. Oxid. Med. Cell. Longev. 2020, 2020, 7901270. [Google Scholar] [CrossRef] [Green Version]
- Glickman, R.D. Ultraviolet phototoxicity to the retina. Eye Contact Lens 2011, 37, 196–205. [Google Scholar] [CrossRef]
- Totan, Y.; Yağcı, R.; Bardak, Y.; Özyurt, H.; Kendir, F.; Yılmaz, G.; Şahin, Ş.; Şahin Tığ, U. Oxidative macromolecular damage in age-related macular degeneration. Curr. Eye Res. 2009, 34, 1089–1093. [Google Scholar] [CrossRef]
- Bhutto, I.A.; Baba, T.; Merges, C.; McLeod, D.S.; Lutty, G.A. Low nitric oxide synthases (NOSs) in eyes with age-related macular degeneration (AMD). Exp. Eye Res. 2010, 90, 155–167. [Google Scholar] [CrossRef] [Green Version]
- Ruan, Y.; Jiang, S.; Gericke, A. Age-related macular degeneration: Role of oxidative stress and blood vessels. Int. J. Mol. Sci. 2021, 22, 1296. [Google Scholar] [CrossRef]
- Toma, C.; De Cillà, S.; Palumbo, A.; Garhwal, D.P.; Grossini, E. Oxidative and nitrosative stress in age-related macular degeneration: A review of their role in different stages of disease. Antioxidants 2021, 10, 653. [Google Scholar] [CrossRef] [PubMed]
- Golestaneh, N.; Chu, Y.; Cheng, S.K.; Cao, H.; Poliakov, E.; Berinstein, D.M. Repressed SIRT1/PGC-1α pathway and mitochondrial disintegration in iPSC-derived RPE disease model of age-related macular degeneration. J. Transl. Med. 2016, 14, 344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imamura, Y.; Noda, S.; Hashizume, K.; Shinoda, K.; Yamaguchi, M.; Uchiyama, S.; Shimizu, T.; Mizushima, Y.; Shirasawa, T.; Tsubota, K. Drusen, choroidal neovascularization, and retinal pigment epithelium dysfunction in SOD1-deficient mice: A model of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2006, 103, 11282–11287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Justilien, V.; Pang, J.-J.; Renganathan, K.; Zhan, X.; Crabb, J.W.; Kim, S.R.; Sparrow, J.R.; Hauswirth, W.W.; Lewin, A.S. SOD2 knockdown mouse model of early AMD. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4407–4420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Chen, Y.; Wang, J.; Sternberg, P.; Freeman, M.L.; Grossniklaus, H.E.; Cai, J. Age-related retinopathy in NRF2-deficient mice. PLoS ONE 2011, 6, e19456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, D.Y.; Frank, S.I.; Fitch, T.C.; Karg, M.M.; Butcher, E.R.; Nnuji-John, E.; Kim, L.A.; Saint-Geniez, M. Dimethyl Fumarate Blocks Tumor Necrosis Factor-Alpha-Driven Inflammation and Metabolic Rewiring in the Retinal Pigment Epithelium. Front. Mol. Neurosci. 2022, 15, 896786. [Google Scholar] [CrossRef]
- Shimizu, H.; Takayama, K.; Yamada, K.; Suzumura, A.; Sato, T.; Nishio, Y.; Ito, M.; Ushida, H.; Nishiguchi, K.M.; Takeuchi, M.; et al. Dimethyl Fumarate Protects Retinal Pigment Epithelium from Blue Light-Induced Oxidative Damage via the Nrf2 Pathway. Antioxidants 2023, 12, 45. [Google Scholar] [CrossRef]
- Ishibashi, T.; Murata, T.; Hangai, M.; Nagai, R.; Horiuchi, S.; Lopez, P.F.; Hinton, D.R.; Ryan, S.J. Advanced glycation end products in age-related macular degeneration. Arch. Ophthalmol. 1998, 116, 1629–1632. [Google Scholar] [CrossRef]
- Kandarakis, S.A.; Piperi, C.; Topouzis, F.; Papavassiliou, A.G. Emerging role of advanced glycation-end products (AGEs) in the pathobiology of eye diseases. Prog. Retin. Eye Res. 2014, 42, 85–102. [Google Scholar] [CrossRef]
- Crabb, J.W.; Miyagi, M.; Gu, X.; Shadrach, K.; West, K.A.; Sakaguchi, H.; Kamei, M.; Hasan, A.; Yan, L.; Rayborn, M.E. Drusen proteome analysis: An approach to the etiology of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 14682–14687. [Google Scholar] [CrossRef] [Green Version]
- Ardeljan, D.; Tuo, J.; Chan, C.-C. Carboxyethylpyrrole plasma biomarkers in age-related macular degeneration. Drugs Future 2011, 36, 712. [Google Scholar] [CrossRef] [Green Version]
- Balaiya, S.; Murthy, R.K.; Brar, V.S.; Chalam, K.V. Evaluation of ultraviolet light toxicity on cultured retinal pigment epithelial and retinal ganglion cells. Clin. Ophthalmol. 2010, 4, 33. [Google Scholar] [PubMed] [Green Version]
- Tong, Y.; Zhang, Z.; Wang, S. Role of Mitochondria in Retinal Pigment Epithelial Aging and Degeneration. Front. Aging 2022, 3, 926627. [Google Scholar] [CrossRef]
- Alaimo, A.; Liñares, G.G.; Bujjamer, J.M.; Gorojod, R.M.; Alcon, S.P.; Martínez, J.H.; Baldessari, A.; Grecco, H.E.; Kotler, M.L. Toxicity of blue led light and A2E is associated to mitochondrial dynamics impairment in ARPE-19 cells: Implications for age-related macular degeneration. Arch. Toxicol. 2019, 93, 1401–1415. [Google Scholar] [CrossRef]
- Tao, J.-X.; Zhou, W.-C.; Zhu, X.-G. Mitochondria as potential targets and initiators of the blue light hazard to the retina. Oxid. Med. Cell. Longev. 2019, 2019, 6435364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulton, M.; Różanowska, M.; Różanowski, B. Retinal photodamage. J. Photochem. Photobiol. B Biol. 2001, 64, 144–161. [Google Scholar] [CrossRef] [PubMed]
- Godley, B.F.; Shamsi, F.A.; Liang, F.-Q.; Jarrett, S.G.; Davies, S.; Boulton, M. Blue light induces mitochondrial DNA damage and free radical production in epithelial cells. J. Biol. Chem. 2005, 280, 21061–21066. [Google Scholar] [CrossRef] [Green Version]
- Lowe, G.M.; Vlismas, K.; Young, A.J. Carotenoids as prooxidants? Mol. Asp. Med. 2003, 24, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Roehlecke, C.; Schumann, U.; Ader, M.; Brunssen, C.; Bramke, S.; Morawietz, H.; Funk, R.H. Stress reaction in outer segments of photoreceptors after blue light irradiation. PLoS ONE 2013, 8, e71570. [Google Scholar] [CrossRef]
- Thornton, J.; Edwards, R.; Mitchell, P.; Harrison, R.; Buchan, I.; Kelly, S.P. Smoking and age-related macular degeneration: A review of association. Eye 2005, 19, 935–944. [Google Scholar] [CrossRef]
- Altuntaş, I.; Dane, Ş.; Gümüştekin, K. Effects of cigarette smoking on lipid peroxidation. J. Basic Clin. Physiol. Pharmacol. 2002, 13, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, S.G.; Boulton, M.E. Consequences of oxidative stress in age-related macular degeneration. Mol. Asp. Med. 2012, 33, 399–417. [Google Scholar] [CrossRef] [Green Version]
- Seddon, J.M.; Gensler, G.; Milton, R.C.; Klein, M.L.; Rifai, N. Association between C-reactive protein and age-related macular degeneration. JAMA 2004, 291, 704–710. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.W.; Jung, I.H.; Park, E.Y.; Kim, K.H.; Kim, K.; Yeom, J.; Jung, J.; Lee, S.W. Radiation-induced C-reactive protein triggers apoptosis of vascular smooth muscle cells through ROS interfering with the STAT3/Ref-1 complex. J. Cell. Mol. Med. 2022, 26, 2104–2118. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhang, J.; Liu, J.; Sun, L.; Zhao, H.; Lu, Y.; Wang, J.; Li, J. C-reactive protein promotes adhesion of monocytes to endothelial cells via NADPH oxidase-mediated oxidative stress. J. Cell. Biochem. 2012, 113, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Hadzic, S.; Roxlau, E.T.; Fuehler, B.; Janise-Libawski, A.; Wimmer, T.; Lei, B.; Li, S.-W.; Weissmann, N.; Stieger, K. Retinal tissue develops an inflammatory reaction to tobacco smoke and electronic cigarette vapor in mice. J. Mol. Med. 2021, 99, 1459–1469. [Google Scholar] [CrossRef]
- Delcourt, C.; Delyfer, M.-N.; Rougier, M.-B.; Amouyel, P.; Colin, J.; Le Goff, M.; Malet, F.; Dartigues, J.-F.; Lambert, J.-C.; Korobelnik, J.-F. Associations of complement factor H and smoking with early age-related macular degeneration: The ALIENOR study. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5955–5962. [Google Scholar] [CrossRef]
- Mitter, S.K.; Rao, H.V.; Qi, X.; Cai, J.; Sugrue, A.; Dunn, W.A.; Grant, M.B.; Boulton, M.E. Autophagy in the retina: A potential role in age-related macular degeneration. Retin. Degener. Dis. 2012, 723, 83–90. [Google Scholar]
- Hyttinen, J.; Blasiak, J.; Tavi, P.; Kaarniranta, K. Therapeutic potential of PGC-1α in age-related macular degeneration (AMD)–the involvement of mitochondrial quality control, autophagy, and antioxidant response. Expert Opin. Ther. Targets 2021, 25, 773–785. [Google Scholar] [CrossRef]
- Chen, C.-L.; Chen, Y.-H.; Liang, C.-M.; Tai, M.-C.; Lu, D.-W.; Chen, J.-T. Glucosamine-induced autophagy through AMPK–mTOR pathway attenuates lipofuscin-like autofluorescence in human retinal pigment epithelial cells in vitro. Int. J. Mol. Sci. 2018, 19, 1416. [Google Scholar] [CrossRef] [Green Version]
- Sethna, S.; Scott, P.A.; Giese, A.P.; Duncan, T.; Jian, X.; Riazuddin, S.; Randazzo, P.A.; Redmond, T.M.; Bernstein, S.L.; Riazuddin, S. CIB2 regulates mTORC1 signaling and is essential for autophagy and visual function. Nat. Commun. 2021, 12, 3906. [Google Scholar] [CrossRef] [PubMed]
- Riazi-Esfahani, M.; Kuppermann, B.D.; Kenney, M.C. The role of mitochondria in AMD: Current knowledge and future applications. J. Ophthalmic Vis. Res. 2017, 12, 424. [Google Scholar] [PubMed]
- Sridevi Gurubaran, I.; Viiri, J.; Koskela, A.; Hyttinen, J.M.; Paterno, J.J.; Kis, G.; Antal, M.; Urtti, A.; Kauppinen, A.; Felszeghy, S. Mitophagy in the retinal pigment epithelium of dry age-related macular degeneration investigated in the NFE2l2/PGC-1α-/-mouse model. Int. J. Mol. Sci. 2020, 21, 1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyttinen, J.M.; Viiri, J.; Kaarniranta, K.; Błasiak, J. Mitochondrial quality control in AMD: Does mitophagy play a pivotal role? Cell. Mol. Life Sci. 2018, 75, 2991–3008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.-Y.; Bao, X.-L.; Cong, Y.-Y.; Fan, B.; Li, G.-Y. Autophagy in age-related macular degeneration: A regulatory mechanism of oxidative stress. Oxid. Med. Cell. Longev. 2020, 2020, 2896036. [Google Scholar] [CrossRef]
- Zhao, T.; Guo, X.; Sun, Y. Iron accumulation and lipid peroxidation in the aging retina: Implication of ferroptosis in age-related macular degeneration. Aging Dis. 2021, 12, 529. [Google Scholar] [CrossRef]
- Gnana-Prakasam, J.P.; Tawfik, A.; Romej, M.; Ananth, S.; Martin, P.M.; Smith, S.B.; Ganapathy, V. Iron-mediated retinal degeneration in haemojuvelin-knockout mice. Biochem. J. 2012, 441, 599–608. [Google Scholar] [CrossRef]
- Dunaief, J.L. Iron induced oxidative damage as a potential factor in age-related macular degeneration: The Cogan Lecture. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4660–4664. [Google Scholar] [CrossRef] [Green Version]
- Ashok, A.; Chaudhary, S.; Wise, A.S.; Rana, N.A.; McDonald, D.; Kritikos, A.E.; Lindner, E.; Singh, N. Release of Iron-Loaded Ferritin in Sodium Iodate-Induced Model of Age Related Macular Degeneration: An In-Vitro and In-Vivo Study. Antioxidants 2021, 10, 1253. [Google Scholar] [CrossRef]
- Papadopoulos, N.; Martin, J.; Ruan, Q.; Rafique, A.; Rosconi, M.P.; Shi, E.; Pyles, E.A.; Yancopoulos, G.D.; Stahl, N.; Wiegand, S.J. Binding and neutralization of vascular endothelial growth factor (VEGF) and related ligands by VEGF Trap, ranibizumab and bevacizumab. Angiogenesis 2012, 15, 171–185. [Google Scholar] [CrossRef] [Green Version]
- Heier, J.S.; Brown, D.M.; Chong, V.; Korobelnik, J.-F.; Kaiser, P.K.; Nguyen, Q.D.; Kirchhof, B.; Ho, A.; Ogura, Y.; Yancopoulos, G.D. Intravitreal aflibercept (VEGF trap-eye) in wet age-related macular degeneration. Ophthalmology 2012, 119, 2537–2548. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.F.; Maguire, M.G.; Ying, G.; Grunwald, J.E.; Fine, S.L.; Jaffe, G.J. Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N. Engl. J. Med. 2011, 364, 1897–1908. [Google Scholar] [PubMed] [Green Version]
- Mimouni, M.; Meshi, A.; Vainer, I.; Gershoni, A.; Koren, T.; Geffen, N.; Nemet, A.Y.; Segal, O. Bevacizumab dosing every 2 weeks for neovascular age-related macular degeneration refractory to monthly dosing. Jpn. J. Ophthalmol. 2018, 62, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Damico, L.; Shams, N.; Lowman, H.; Kim, R. Development of ranibizumab, an anti–vascular endothelial growth factor antigen binding fragment, as therapy for neovascular age-related macular degeneration. Retina 2006, 26, 859–870. [Google Scholar] [CrossRef]
- Markham, A. Brolucizumab: First approval. Drugs 2019, 79, 1997–2000. [Google Scholar] [CrossRef]
- FDA. Drug Administration Center for Drug Evaluation and Research; Application Number 204042Orig1s000: Summary Review; Food and Drug Administration: Silver Spring, MD, USA, 2017. [Google Scholar]
- Wang, Y.; Wang, V.; Chan, C. The role of anti-inflammatory agents in age-related macular degeneration (AMD) treatment. Eye 2011, 25, 127–139. [Google Scholar] [CrossRef]
- De Guimaraes, T.A.C.; Varela, M.D.; Georgiou, M.; Michaelides, M. Treatments for dry age-related macular degeneration: Therapeutic avenues, clinical trials and future directions. Br. J. Ophthalmol. 2022, 106, 297–304. [Google Scholar] [CrossRef]
- Liao, D.S.; Grossi, F.V.; El Mehdi, D.; Gerber, M.R.; Brown, D.M.; Heier, J.S.; Wykoff, C.C.; Singerman, L.J.; Abraham, P.; Grassmann, F. Complement C3 inhibitor pegcetacoplan for geographic atrophy secondary to age-related macular degeneration: A randomized phase 2 trial. Ophthalmology 2020, 127, 186–195. [Google Scholar] [CrossRef] [Green Version]
- Potilinski, M.C.; Tate, P.S.; Lorenc, V.E.; Gallo, J.E. New insights into oxidative stress and immune mechanisms involved in age-related macular degeneration tackled by novel therapies. Neuropharmacology 2021, 188, 108513. [Google Scholar] [CrossRef]
- Dziedziak, J.; Kasarełło, K.; Cudnoch-Jędrzejewska, A. Dietary antioxidants in age-related macular degeneration and glaucoma. Antioxidants 2021, 10, 1743. [Google Scholar] [CrossRef]
- Age-Related Eye Disease Study Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch. Ophthalmol. 2001, 119, 1417–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Age-Related Eye Disease Study 2 (AREDS2) Research Group. Lutein+ zeaxanthin and omega-3 fatty acids for age-related macular degeneration: The Age-Related Eye Disease Study 2 (AREDS2) randomized clinical trial. JAMA 2013, 309, 2005–2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, G.; Olson, C.G.; Euritt, C.P.; Koulen, P. Molecular Mechanisms Underlying the Therapeutic Role of Vitamin E in Age-Related Macular Degeneration. Front. Neurosci. 2022, 16, 890021. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Igarashi, K.; Uchihara, T.; Jishage, K.-i.; Tomita, H.; Inaba, A.; Li, Y.; Arita, M.; Suzuki, H.; Mizusawa, H. Delayed-onset ataxia in mice lacking α-tocopherol transfer protein: Model for neuronal degeneration caused by chronic oxidative stress. Proc. Natl. Acad. Sci. USA 2001, 98, 15185–15190. [Google Scholar] [CrossRef] [Green Version]
- Kirchhof, B. Strategies to influence PVR development. Graefe’s Arch. Clin. Exp. Ophthalmol. 2004, 242, 699–703. [Google Scholar] [CrossRef]
- Mudhar, H.S. A brief review of the histopathology of proliferative vitreoretinopathy (PVR). Eye 2020, 34, 246–250. [Google Scholar] [CrossRef]
- Tseng, W.; Cortez, R.T.; Ramirez, G.; Stinnett, S.; Jaffe, G.J. Prevalence and risk factors for proliferative vitreoretinopathy in eyes with rhegmatogenous retinal detachment but no previous vitreoretinal surgery. Am. J. Ophthalmol. 2004, 137, 1105–1115. [Google Scholar] [CrossRef]
- Scott, I.U.; Flynn, H.W., Jr.; Murray, T.G.; Feuer, W.J. Outcomes of surgery for retinal detachment associated with proliferative vitreoretinopathy using perfluoro-n-octane: A multicenter study. Am. J. Ophthalmol. 2003, 136, 454–463. [Google Scholar] [CrossRef]
- Verdejo, C.; Marco, P.; Renau-Piqueras, J.; Pinazo-Duran, M.D. Lipid peroxidation in proliferative vitreoretinopathies. Eye 1999, 13, 183–188. [Google Scholar] [CrossRef] [Green Version]
- Cederlund, M.; Ghosh, F.; Arner, K.; Andreasson, S.; Akerstrom, B. Vitreous levels of oxidative stress biomarkers and the radical-scavenger alpha1-microglobulin/A1M in human rhegmatogenous retinal detachment. Graefe’s Arch. Clin. Exp. Ophthalmol. 2013, 251, 725–732. [Google Scholar] [CrossRef] [Green Version]
- Maeno, A.; Suzuki, Y.; Adachi, K.; Takahashi, S.; Yokoi, Y.; Nakazawa, M. Characterization of the biological antioxidant potential in the vitreous fluid from patients with rhegmatogenous retinal detachment. Acta Ophthalmol. 2016, 94, e515–e516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietras-Baczewska, A.; Nowomiejska, K.; Brzozowska, A.; Toro, M.D.; Załuska, W.; Sztanke, M.; Sztanke, K.; Rejdak, R. Antioxidant Status in the Vitreous of Eyes with Rhegmatogenous Retinal Detachment with and without Proliferative Vitreoretinopathy, Macular Hole and Epiretinal Membrane. Life 2021, 11, 453. [Google Scholar] [CrossRef]
- Cicik, E.; Tekin, H.; Akar, S.; Ekmekci, O.B.; Donma, O.; Koldas, L.; Ozkan, S. Interleukin-8, nitric oxide and glutathione status in proliferative vitreoretinopathy and proliferative diabetic retinopathy. Ophthalmic Res. 2003, 35, 251–255. [Google Scholar] [CrossRef]
- Augustin, A.J.; Spitznas, M.; Koch, F.; Grus, F.; Boker, T. Indicators of oxidative tissue damage and inflammatory activity in epiretinal membranes of proliferative diabetic retinopathy, proliferative vitreoretinopathy and macular pucker. Ger. J. Ophthalmol. 1995, 4, 47–51. [Google Scholar] [PubMed]
- Delgado-Tirado, S.; Amarnani, D.; Zhao, G.; Rossin, E.J.; Eliott, D.; Miller, J.B.; Greene, W.A.; Ramos, L.; Arevalo-Alquichire, S.; Leyton-Cifuentes, D.; et al. Topical delivery of a small molecule RUNX1 transcription factor inhibitor for the treatment of proliferative vitreoretinopathy. Sci. Rep. 2020, 10, 20554. [Google Scholar] [CrossRef] [PubMed]
- Shu, D.Y.; Butcher, E.; Saint-Geniez, M. EMT and EndMT: Emerging Roles in Age-Related Macular Degeneration. Int. J. Mol. Sci. 2020, 21, 4271. [Google Scholar] [CrossRef]
- Shu, D.Y.; Lovicu, F.J. Myofibroblast transdifferentiation: The dark force in ocular wound healing and fibrosis. Prog. Retin. Eye Res. 2017, 60, 44–65. [Google Scholar] [CrossRef]
- Roybal, C.N.; Velez, G.; Toral, M.A.; Tsang, S.H.; Bassuk, A.G.; Mahajan, V.B. Personalized Proteomics in Proliferative Vitreoretinopathy Implicate Hematopoietic Cell Recruitment and mTOR as a Therapeutic Target. Am. J. Ophthalmol. 2018, 186, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.H.; Lee, J.-J.; Wu, P.-C.; Kuo, H.-K.; Kuo, Y.-H.; Huang, H.-M. Oxidative stress enhanced the transforming growth factor-β2-induced epithelial-mesenchymal transition through chemokine ligand 1 on ARPE-19 cell. Sci. Rep. 2020, 10, 4000. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Kim, Y.S.; Kim, J.H.; Jang, H.Y.; Ly, D.D.; Das, R.; Park, K.S. Activation of ERK1/2-mTORC1-NOX4 mediates TGF-beta1-induced epithelial-mesenchymal transition and fibrosis in retinal pigment epithelial cells. Biochem. Biophys. Res. Commun. 2020, 529, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Shi, D.P.; Chu, W.J.; Yang, L.L.; Xu, H.F. LRG1 promotes epithelial-mesenchymal transition of retinal pigment epithelium cells by activating NOX4. Int. J. Ophthalmol. 2021, 14, 349–355. [Google Scholar] [CrossRef]
- Lee, S.J.; Kim, S.J.; Jo, D.H.; Park, K.S.; Kim, J.H. Blockade of mTORC1-NOX signaling pathway inhibits TGF-beta1-mediated senescence-like structural alterations of the retinal pigment epithelium. FASEB J. 2021, 35, e21403. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, J.; Wang, Q.; Xing, Y.; Tan, Z.; Kang, Q. Novel NADPH oxidase inhibitor VAS2870 suppresses TGF-beta-dependent epithelial-to-mesenchymal transition in retinal pigment epithelial cells. Int. J. Mol. Med. 2018, 42, 123–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wada, I.; Sreekumar, P.G.; Spee, C.; MacKay, A.J.; Ip, M.; Kannan, R. Mechanisms of Epithelial-Mesenchymal Transition and Prevention of Dispase-Induced PVR by Delivery of an Antioxidant alphaB Crystallin Peptide. Antioxidants 2022, 11, 2080. [Google Scholar] [CrossRef]
- Cai, W.; Yu, D.; Fan, J.; Liang, X.; Jin, H.; Liu, C.; Zhu, M.; Shen, T.; Zhang, R.; Hu, W.; et al. Quercetin inhibits transforming growth factor beta1-induced epithelial-mesenchymal transition in human retinal pigment epithelial cells via the Smad pathway. Drug Des. Devel. Ther. 2018, 12, 4149–4161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, D.Y.; Butcher, E.R.; Saint-Geniez, M. Suppression of PGC-1alpha Drives Metabolic Dysfunction in TGFbeta2-Induced EMT of Retinal Pigment Epithelial Cells. Int. J. Mol. Sci. 2021, 22, 4701. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Zhang, Y.; Chen, J.; Wu, L.D.; Chen, M.L.; Chen, C.M.; Xu, Q.H. Artesunate inhibits the development of PVR by suppressing the TGF-beta/Smad signaling pathway. Exp. Eye Res. 2021, 213, 108859. [Google Scholar] [CrossRef] [PubMed]
- Cheung, N.; Mitchell, P.; Wong, T.Y. Diabetic retinopathy. Lancet 2010, 376, 124–136. [Google Scholar] [CrossRef]
- Frank, R.N. Diabetic Retinopathy. N. Engl. J. Med. 2004, 350, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Lieth, E.; Barber, A.J.; Xu, B.; Dice, C.; Ratz, M.J.; Tanase, D.; Strother, J.M. Glial reactivity and impaired glutamate metabolism in short-term experimental diabetic retinopathy. Penn State Retina Research Group. Diabetes 1998, 47, 815–820. [Google Scholar] [CrossRef] [PubMed]
- Hombrebueno, J.R.; Chen, M.; Penalva, R.G.; Xu, H. Loss of synaptic connectivity, particularly in second order neurons is a key feature of diabetic retinal neuropathy in the Ins2Akita mouse. PLoS ONE 2014, 9, e97970. [Google Scholar] [CrossRef] [PubMed]
- Barber, A.J.; Lieth, E.; Khin, S.A.; Antonetti, D.A.; Buchanan, A.G.; Gardner, T.W. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J. Clin. Investig. 1998, 102, 783–791. [Google Scholar] [CrossRef] [Green Version]
- Russell, J.W.; Sullivan, K.A.; Windebank, A.J.; Herrmann, D.N.; Feldman, E.L. Neurons undergo apoptosis in animal and cell culture models of diabetes. Neurobiol. Dis. 1999, 6, 347–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, H.-Y.; Green, W.R.; Tso, M.O.M. Microglial Activation in Human Diabetic Retinopathy. Arch. Ophthalmol. 2008, 126, 227–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diabetes, C.; Complications Trial Research, G.; Nathan, D.M.; Genuth, S.; Lachin, J.; Cleary, P.; Crofford, O.; Davis, M.; Rand, L.; Siebert, C. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 1993, 329, 977–986. [Google Scholar] [CrossRef]
- King, P.; Peacock, I.; Donnelly, R. The UK prospective diabetes study (UKPDS): Clinical and therapeutic implications for type 2 diabetes. Br. J. Clin. Pharmacol. 1999, 48, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. The Pathobiology of Diabetic Complications. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders, L.H.; Timothy Greenamyre, J. Oxidative damage to macromolecules in human Parkinson disease and the rotenone model. Free Radic. Biol. Med. 2013, 62, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Forman, H.J.; Maiorino, M.; Ursini, F. Signaling functions of reactive oxygen species. Biochemistry 2010, 49, 835–842. [Google Scholar] [CrossRef] [Green Version]
- Kowluru, R.A. Effect of Reinstitution of Good Glycemic Control on Retinal Oxidative Stress and Nitrative Stress in Diabetic Rats. Diabetes 2003, 52, 818–823. [Google Scholar] [CrossRef] [Green Version]
- Rohowetz, L.J.; Kraus, J.G.; Koulen, P. Reactive Oxygen Species-Mediated Damage of Retinal Neurons: Drug Development Targets for Therapies of Chronic Neurodegeneration of the Retina. Int. J. Mol. Sci. 2018, 19, 3362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Medina, J.; Zanon-Moreno, V.; Pinazo-Duran, M.; Foulquie-Moreno, E.; Rubio-Velazquez, E.; Casaroli-Marano, R.; Del-Rio-Vellosillo, M. Oxidative Stress in Diabetic Retinopathy, 2nd ed.; Preedy, V.R., Ed.; Academic Press: Cambridge, MA, USA, 2020; p. 52. [Google Scholar]
- Field, M.G.; Yang, D.; Bian, Z.-M.; Petty, H.R.; Elner, V.M. Retinal flavoprotein fluorescence correlates with mitochondrial stress, apoptosis, and chemokine expression. Exp. Eye Res. 2011, 93, 548–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, D.C. Diseases Of The Mitochondrial DNA. Annu. Rev. Biochem. 1992, 61, 1175–1212. [Google Scholar] [CrossRef] [PubMed]
- Trumpower, B.L. The protonmotive Q cycle. Energy transduction by coupling of proton translocation to electron transfer by the cytochrome bc1 complex. J. Biol. Chem. 1990, 265, 11409–11412. [Google Scholar] [CrossRef] [PubMed]
- Du, X.L.; Edelstein, D.; Rossetti, L.; Fantus, I.G.; Goldberg, H.; Ziyadeh, F.; Wu, J.; Brownlee, M. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc. Natl. Acad. Sci. USA 2000, 97, 12222–12226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.-I.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.-P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787. [Google Scholar] [CrossRef]
- Du, X.L.; Edelstein, D.; Dimmeler, S.; Ju, Q.; Sui, C.; Brownlee, M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J. Clin. Investig. 2001, 108, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Giugliano, D.; Ceriello, A.; Paolisso, G. Oxidative Stress and Diabetic Vascular Complications. Diabetes Care 1996, 19, 257–267. [Google Scholar] [CrossRef]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacco, F.; Du, X.; Carratú, A.; Gerfen, G.J.; D’Apolito, M.; Giardino, I.; Rasola, A.; Marin, O.; Divakaruni, A.S.; Murphy, A.N.; et al. GLP-1 Cleavage Product Reverses Persistent ROS Generation After Transient Hyperglycemia by Disrupting an ROS-Generating Feedback Loop. Diabetes 2015, 64, 3273–3284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, W.P.; Toro, A.L.; Barber, A.J.; Dennis, M.D. REDD1 Activates a ROS-Generating Feedback Loop in the Retina of Diabetic Mice REDD1 Promotes Oxidative Stress in Retina. Investig. Ophthalmol. Vis. Sci. 2019, 60, 2369–2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ford, E.S.; Mokdad, A.H.; Giles, W.H.; Brown, D.W. The Metabolic Syndrome and Antioxidant Concentrations. Diabetes 2003, 52, 2346–2352. [Google Scholar] [CrossRef] [Green Version]
- Kowluru, R.A.; Kern, T.S.; Engerman, R.L.; Armstrong, D. Abnormalities of Retinal Metabolism in Diabetes or Experimental Galactosemia. III. Effects of Antioxidants. Diabetes 1996, 45, 1233–1237. [Google Scholar] [CrossRef]
- Wohaieb, S.A.; Godin, D.V. Alterations in Free Radical Tissue-Defense Mechanisms in Streptozocin-Induced Diabetes in Rat: Effects of Insulin Treatment. Diabetes 1987, 36, 1014–1018. [Google Scholar] [CrossRef]
- Baynes, J.W.; Thorpe, S.R. Role of oxidative stress in diabetic complications: A new perspective on an old paradigm. Diabetes 1999, 48, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Haskins, K.; Bradley, B.; Powers, K.; Fadok, V.; Flores, S.; Ling, X.; Pugazhenthi, S.; Reusch, J.; Kench, J. Oxidative Stress in Type 1 Diabetes. Ann. N. Y. Acad. Sci. 2003, 1005, 43–54. [Google Scholar] [CrossRef]
- Takahashi, T.; Harris, R.C. Role of endothelial nitric oxide synthase in diabetic nephropathy: Lessons from diabetic eNOS knockout mice. J. Diabetes Res. 2014, 2014, 590541. [Google Scholar] [CrossRef] [Green Version]
- Felaco, M.; Grilli, A.; De Lutiis, M.A.; Patruno, A.; Libertini, N.; Taccardi, A.A.; Di Napoli, P.; Di Giulio, C.; Barbacane, R.; Conti, P. Endothelial Nitric Oxide Synthase (eNOS) Expression and Localization in Healthy and Diabetic Rat Hearts. Ann. Clin. Lab. Sci. 2001, 31, 179–186. [Google Scholar]
- Carmo, A.; Cunha-Vaz, J.G.; Carvalho, A.P.; Lopes, M.C. Nitric Oxide Synthase Activity in Retinas from Non-Insulin-Dependent Diabetic Goto-Kakizaki Rats: Correlation with Blood–Retinal Barrier Permeability. Nitric Oxide 2000, 4, 590–596. [Google Scholar] [CrossRef]
- Martínez, M.C.; Andriantsitohaina, R. Reactive Nitrogen Species: Molecular Mechanisms and Potential Significance in Health and Disease. Antioxid. Redox. Signal. 2008, 11, 669–702. [Google Scholar] [CrossRef] [PubMed]
- Afanas’ev, I. Signaling of reactive oxygen and nitrogen species in Diabetes mellitus. Oxid. Med. Cell. Longev. 2010, 3, 361–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Stockklauser-Färber, K.; Rösen, P. Generation of reactive oxygen intermediates, activation of NF-κB, and induction of apoptosis in human endothelial cells by glucose: Role of nitric oxide synthase? Free Radic. Biol. Med. 1999, 27, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Chough, E.; Daley, J.; Oates, P.; Tornheim, K.; Ruderman, N.B.; Keaney, J.F. Hyperglycemia increases endothelial superoxide that impairs smooth muscle cell Na+-K+-ATPase activity. Am. J. Physiol. Cell Physiol. 2002, 282, C560–C566. [Google Scholar] [CrossRef]
- Maritim, A.C.; Sanders, R.A.; Watkins Iii, J.B. Diabetes, oxidative stress, and antioxidants: A review. J. Biochem. Mol. Toxicol. 2003, 17, 24–38. [Google Scholar] [CrossRef]
- Golbidi, S.; Alireza Ebadi, S.; Laher, I. Antioxidants in the Treatment of Diabetes. Curr. Diabetes Rev. 2011, 7, 106–125. [Google Scholar] [CrossRef]
- Sheikh-Ali, M.; Chehade, J.M.; Mooradian, A.D. The Antioxidant Paradox in Diabetes Mellitus. Am. J. Ther. 2011, 18, 266–278. [Google Scholar] [CrossRef]
- Miller, W.P.; Sunilkumar, S.; Giordano, J.F.; Toro, A.L.; Barber, A.J.; Dennis, M.D. The stress response protein REDD1 promotes diabetes-induced oxidative stress in the retina by Keap1-independent Nrf2 degradation. J. Biol. Chem. 2020, 295, 7350–7361. [Google Scholar] [CrossRef] [Green Version]
- Marra, G.; Cotroneo, P.; Pitocco, D.; Manto, A.; Di Leo, M.A.S.; Ruotolo, V.; Caputo, S.; Giardina, B.; Ghirlanda, G.; Santini, S.A. Early Increase of Oxidative Stress and Reduced Antioxidant Defenses in Patients With Uncomplicated Type 1 Diabetes. Diabetes Care 2002, 25, 370–375. [Google Scholar] [CrossRef] [Green Version]
- Martín-Gallán, P.; Carrascosa, A.; Gussinye, M.; Domínguez, C. Estimation of lipoperoxidative damage and antioxidant status in diabetic children: Relationship with individual antioxidants. Free Radic. Res. 2005, 39, 933–942. [Google Scholar] [CrossRef]
- Bhatia, S.; Shukla, R.; Venkata Madhu, S.; Kaur Gambhir, J.; Madhava Prabhu, K. Antioxidant status, lipid peroxidation and nitric oxide end products in patients of type 2 diabetes mellitus with nephropathy. Clin. Biochem. 2003, 36, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yu, H.; Pan, H.; Zhou, X.; Ruan, Q.; Kong, D.; Chu, Z.; Li, H.; Huang, J.; Huang, X.; et al. Nrf2 Suppression Delays Diabetic Wound Healing Through Sustained Oxidative Stress and Inflammation. Front. Pharmacol. 2019, 10, 1099. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Chen, X.; Niu, C.; Huang, X.; An, N.; Sun, J.; Huang, S.; Ye, W.; Li, S.; Shen, Y.; et al. Baicalin alleviates hyperglycemia-induced endothelial impairment via Nrf2. J. Endocrinol. 2019, 240, 81–98. [Google Scholar] [CrossRef] [PubMed]
- Shivarudrappa, A.H.; Ponesakki, G. Lutein reverses hyperglycemia-mediated blockage of Nrf2 translocation by modulating the activation of intracellular protein kinases in retinal pigment epithelial (ARPE-19) cells. J. Cell Commun. Signal. 2020, 14, 207–221. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Wong-Riley, M.T. Energy metabolism of the visual system. Eye Brain 2010, 2, 99–116. [Google Scholar] [CrossRef] [Green Version]
- Donato, L.; Scimone, C.; Rinaldi, C.; Aragona, P.; Briuglia, S.; D’Ascola, A.; D’Angelo, R.; Sidoti, A. Stargardt Phenotype Associated With Two ELOVL4 Promoter Variants and ELOVL4 Downregulation: New Possible Perspective to Etiopathogenesis? Investig. Ophthalmol. Vis. Sci. 2018, 59, 843–857. [Google Scholar] [CrossRef] [Green Version]
- Kowluru, R.A.; Abbas, S.N. Diabetes-Induced Mitochondrial Dysfunction in the Retina. Investig. Ophthalmol. Vis. Sci. 2003, 44, 5327–5334. [Google Scholar] [CrossRef] [Green Version]
- Ellis, E.A.; Guberski, D.L.; Somogyi-Mann, M.; Grant, M.B. Increased H2O2, vascular endothelial growth factor and receptors in the retina of the BBZ/WOR diabetic rat. Free Radic. Biol. Med. 2000, 28, 91–101. [Google Scholar] [CrossRef]
- Miller, W.P.; Sha, C.M.; Sunilkumar, S.; Toro, A.L.; VanCleave, A.M.; Kimball, S.R.; Dokholyan, N.V.; Dennis, M.D. Activation of Disulfide Redox Switch in REDD1 Promotes Oxidative Stress Under Hyperglycemic Conditions. Diabetes 2022, 71, 2764–2776. [Google Scholar] [CrossRef]
- Zheng, L.; Du, Y.; Miller, C.; Gubitosi-Klug, R.A.; Kern, T.S.; Ball, S.; Berkowitz, B.A. Critical role of inducible nitric oxide synthase in degeneration of retinal capillaries in mice with streptozotocin-induced diabetes. Diabetologia 2007, 50, 1987–1996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Y.; Smith, M.A.; Miller, C.M.; Kern, T.S. Diabetes-induced nitrative stress in the retina, and correction by aminoguanidine. J. Neurochem. 2002, 80, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Kanwar, M.; Kennedy, A. Metabolic memory phenomenon and accumulation of peroxynitrite in retinal capillaries. Exp. Diabetes Res. 2007, 2007, 21976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhar, S.S.; Liang, H.L.; Wong-Riley, M.T. Transcriptional coupling of synaptic transmission and energy metabolism: Role of nuclear respiratory factor 1 in co-regulating neuronal nitric oxide synthase and cytochrome c oxidase genes in neurons. Biochim. Biophys. Acta 2009, 1793, 1604–1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, M.; Barber, A.J.; Antonetti, D.A.; LaNoue, K.F.; Robinson, K.A.; Buse, M.G.; Gardner, T.W. Excessive Hexosamines Block the Neuroprotective Effect of Insulin and Induce Apoptosis in Retinal Neurons. J. Biol. Chem. 2001, 276, 43748–43755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hangai, M.; He, S.; Hoffmann, S.; Lim, J.I.; Ryan, S.J.; Hinton, D.R. Sequential induction of angiogenic growth factors by TNF-alpha in choroidal endothelial cells. J. Neuroimmunol. 2006, 171, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Koppolu, P. Diabetes-induced activation of caspase-3 in retina: Effect of antioxidant therapy. Free Radic. Res. 2002, 36, 993–999. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Koppolu, P.; Chakrabarti, S.; Chen, S. Diabetes-induced activation of nuclear transcriptional factor in the retina, and its inhibition by antioxidants. Free Radic. Res. 2003, 37, 1169–1180. [Google Scholar] [CrossRef]
- Mizutani, M.; Gerhardinger, C.; Lorenzi, M. Müller cell changes in human diabetic retinopathy. Diabetes 1998, 47, 445–449. [Google Scholar] [CrossRef]
- Phipps, J.A.; Fletcher, E.L.; Vingrys, A.J. Paired-flash identification of rod and cone dysfunction in the diabetic rat. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4592–4600. [Google Scholar] [CrossRef] [Green Version]
- Ali, T.K.; Al-Gayyar, M.M.; Matragoon, S.; Pillai, B.A.; Abdelsaid, M.A.; Nussbaum, J.J.; El-Remessy, A.B. Diabetes-induced peroxynitrite impairs the balance of pro-nerve growth factor and nerve growth factor, and causes neurovascular injury. Diabetologia 2011, 54, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.M.; Mohammad, G.; Kowluru, Q.Z.; Renu, A. Diabetic Retinopathy, Superoxide Damage and Antioxidants. Curr. Pharm. Biotechnol. 2011, 12, 352–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Yanoff, M.; Jian, B.; He, Z. Altered mRNA Levels of Antioxidant Enzymes in Pre-Apoptotic Pericytes From Human Diabetic Retinas. Cell. Mol. Biol. 1999, 45, 59–66. [Google Scholar] [PubMed]
- Zhong, Q.; Mishra, M.; Kowluru, R.A. Transcription Factor Nrf2-Mediated Antioxidant Defense System in the Development of Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3941–3948. [Google Scholar] [CrossRef]
- Yue, K.K.M.; Chung, W.-S.; Leung, A.W.N.; Cheng, C.H.K. Redox changes precede the occurrence of oxidative stress in eyes and aorta, but not in kidneys of diabetic rats. Life Sci. 2003, 73, 2557–2570. [Google Scholar] [CrossRef]
- Miranda, M.; Muriach, M.; Roma, J.; Bosch-Morell, F.; Genovés, J.M.; Barcia, J.; Araiz, J.; Díaz-Llopis, M.; Romero, F.J. Oxidative Stress In a Model of Experimental Diabetic Retinopathy: The Utility of Peroxynitrite Scavengers. Arch. Soc. Esp. Oftalmol. 2006, 81, 27–32. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.H.; Jiang, D.Y.; Tang, L.S. Advanced glycation end-products induce apoptosis involving the signaling pathways of oxidative stress in bovine retinal pericytes. Life Sci. 2006, 79, 1040–1048. [Google Scholar] [CrossRef]
- Kowluru, R.A. Effect of advanced glycation end products on accelerated apoptosis of retinal capillary cells under in vitro conditions. Life Sci. 2005, 76, 1051–1060. [Google Scholar] [CrossRef]
- Millen, A.E.; Klein, R.; Folsom, A.R.; Stevens, J.; Palta, M.; Mares, J.A. Relation between intake of vitamins C and E and risk of diabetic retinopathy in the Atherosclerosis Risk in Communities Study. Am. J. Clin. Nutr. 2004, 79, 865–873. [Google Scholar] [CrossRef] [Green Version]
- Mayer-Davis, E.J.; Bell, R.A.; Reboussin, B.A.; Rushing, J.; Marshall, J.A.; Hamman, R.F. Antioxidant nutrient intake and diabetic retinopathy: The San Luis Valley Diabetes Study. Ophthalmology 1998, 105, 2264–2270. [Google Scholar] [CrossRef]
- Millen, A.E.; Gruber, M.; Klein, R.; Klein, B.E.; Palta, M.; Mares, J.A. Relations of serum ascorbic acid and alpha-tocopherol to diabetic retinopathy in the Third National Health and Nutrition Examination Survey. Am. J. Epidemiol. 2003, 158, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Tham, Y.C.; Li, X.; Wong, T.Y.; Quigley, H.A.; Aung, T.; Cheng, C.Y. Global prevalence of glaucoma and projections of glaucoma burden through 2040: A systematic review and meta-analysis. Ophthalmology 2014, 121, 2081–2090. [Google Scholar] [CrossRef]
- Coleman, A.L.; Miglior, S. Risk factors for glaucoma onset and progression. Surv. Ophthalmol. 2008, 53 (Suppl. S1), S3–S10. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.K.; Kee, C. Population-based glaucoma prevalence studies in Asians. Surv. Ophthalmol. 2014, 59, 434–447. [Google Scholar] [CrossRef] [PubMed]
- Esporcatte, B.L.; Tavares, I.M. Normal-tension glaucoma: An update. Arq. Bras. Oftalmol. 2016, 79, 270–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, S.M.; Lerner, S.F.; Brunzini, R.; Evelson, P.A.; Llesuy, S.F. Oxidative stress markers in aqueous humor of glaucoma patients. Am. J. Ophthalmol. 2004, 137, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Tanito, M.; Kaidzu, S.; Takai, Y.; Ohira, A. Correlation between Systemic Oxidative Stress and Intraocular Pressure Level. PLoS ONE 2015, 10, e0133582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Van Bergen, N.J.; Kong, G.Y.; Chrysostomou, V.; Waugh, H.S.; O’Neill, E.C.; Crowston, J.G.; Trounce, I.A. Mitochondrial dysfunction in glaucoma and emerging bioenergetic therapies. Exp. Eye Res. 2011, 93, 204–212. [Google Scholar] [CrossRef]
- Casson, R.J.; Chidlow, G.; Wood, J.P.M. Estimate of the adenosine triphosphate requirement of human retinal ganglion cells. Clin. Exp. Ophthalmol. 2019, 47, 683–684. [Google Scholar] [CrossRef]
- Nita, M.; Grzybowski, A. The Role of the Reactive Oxygen Species and Oxidative Stress in the Pathomechanism of the Age-Related Ocular Diseases and Other Pathologies of the Anterior and Posterior Eye Segments in Adults. Oxid. Med. Cell Longev. 2016, 2016, 3164734. [Google Scholar] [CrossRef] [Green Version]
- Baudouin, C.; Kolko, M.; Melik-Parsadaniantz, S.; Messmer, E.M. Inflammation in Glaucoma: From the back to the front of the eye, and beyond. Prog. Retin. Eye Res. 2021, 83, 100916. [Google Scholar] [CrossRef]
- Tsai, J. Should we measure (and treat) ocular perfusion pressure in glaucoma patients? Glaucoma Today 2009, 5, 31–35. [Google Scholar]
- Zhao, J.; Wang, S.; Zhong, W.; Yang, B.; Sun, L.; Zheng, Y. Oxidative stress in the trabecular meshwork (Review). Int. J. Mol. Med. 2016, 38, 995–1002. [Google Scholar] [CrossRef] [Green Version]
- Mozaffarieh, M.; Grieshaber, M.C.; Flammer, J. Oxygen and blood flow: Players in the pathogenesis of glaucoma. Mol. Vis. 2008, 14, 224–233. [Google Scholar]
- Iyama, T.; Wilson, D.M., III. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair 2013, 12, 620–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [Green Version]
- Cuchra, M.; Markiewicz, L.; Mucha, B.; Pytel, D.; Szymanek, K.; Szemraj, J.; Szaflik, J.; Szaflik, J.P.; Majsterek, I. The role of base excision repair in the development of primary open angle glaucoma in the Polish population. Mutat. Res. 2015, 778, 26–40. [Google Scholar] [CrossRef]
- Sacca, S.C.; Pascotto, A.; Camicione, P.; Capris, P.; Izzotti, A. Oxidative DNA damage in the human trabecular meshwork: Clinical correlation in patients with primary open-angle glaucoma. Arch. Ophthalmol. 2005, 123, 458–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohanty, K.; Dada, R.; Dada, T. Oxidative DNA damage and reduced expression of DNA repair genes: Role in primary open angle glaucoma (POAG). Ophthalmic Genet. 2017, 38, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Caballero, M.; Liton, P.B.; Epstein, D.L.; Gonzalez, P. Proteasome inhibition by chronic oxidative stress in human trabecular meshwork cells. Biochem. Biophys. Res. Commun. 2003, 308, 346–352. [Google Scholar] [CrossRef]
- Fatma, N.; Kubo, E.; Toris, C.B.; Stamer, W.D.; Camras, C.B.; Singh, D.P. PRDX6 attenuates oxidative stress- and TGFbeta-induced abnormalities of human trabecular meshwork cells. Free Radic. Res. 2009, 43, 783–795. [Google Scholar] [CrossRef] [Green Version]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef] [PubMed]
- Sreekumar, P.G.; Hinton, D.R.; Kannan, R. The Emerging Role of Senescence in Ocular Disease. Oxid. Med. Cell Longev. 2020, 2020, 2583601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flammer, J.; Konieczka, K.; Flammer, A.J. The primary vascular dysregulation syndrome: Implications for eye diseases. EPMA J. 2013, 4, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, P.A.; Harder, J.M.; Foxworth, N.E.; Cochran, K.E.; Philip, V.M.; Porciatti, V.; Smithies, O.; John, S.W. Vitamin B(3) modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science 2017, 355, 756–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, T.H.; Romano, G.L.; Amato, R.; Porciatti, V. Nicotinamide-Rich Diet in DBA/2J Mice Preserves Retinal Ganglion Cell Metabolic Function as Assessed by PERG Adaptation to Flicker. Nutrients 2020, 12, 1910. [Google Scholar] [CrossRef] [PubMed]
- Kouassi Nzoughet, J.; Chao de la Barca, J.M.; Guehlouz, K.; Leruez, S.; Coulbault, L.; Allouche, S.; Bocca, C.; Muller, J.; Amati-Bonneau, P.; Gohier, P.; et al. Nicotinamide Deficiency in Primary Open-Angle Glaucoma. Investig. Ophthalmol. Vis. Sci. 2019, 60, 2509–2514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinon, F. Signaling by ROS drives inflammasome activation. Eur. J. Immunol. 2010, 40, 616–619. [Google Scholar] [CrossRef]
- Ji, L.L.; Yeo, D. Oxidative stress: An evolving definition. Fac. Rev. 2021, 10, 13. [Google Scholar] [CrossRef]
- Canizales, L.; Rodriguez, L.; Rivera, C.; Martinez, A.; Mendez, F.; Castillo, A. Low-level expression of SOD1 in peripheral blood samples of patients diagnosed with primary open-angle glaucoma. Biomark. Med. 2016, 10, 1218–1223. [Google Scholar] [CrossRef]
- Hashizume, K.; Hirasawa, M.; Imamura, Y.; Noda, S.; Shimizu, T.; Shinoda, K.; Kurihara, T.; Noda, K.; Ozawa, Y.; Ishida, S.; et al. Retinal dysfunction and progressive retinal cell death in SOD1-deficient mice. Am. J. Pathol. 2008, 172, 1325–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NAD(P)H oxidase: Role in cardiovascular biology and disease. Circ. Res. 2000, 86, 494–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridovich, I. Mitochondria: Are they the seat of senescence? Aging Cell 2004, 3, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Chidlow, G.; Wood, J.P.M.; Casson, R.J. Investigations into Hypoxia and Oxidative Stress at the Optic Nerve Head in a Rat Model of Glaucoma. Front. Neurosci. 2017, 11, 478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan Gaskin, J.C.; Shah, M.H.; Chan, E.C. Oxidative Stress and the Role of NADPH Oxidase in Glaucoma. Antioxidants 2021, 10, 238. [Google Scholar] [CrossRef] [PubMed]
- Nisimoto, Y.; Diebold, B.A.; Cosentino-Gomes, D.; Lambeth, J.D. Nox4: A hydrogen peroxide-generating oxygen sensor. Biochemistry 2014, 53, 5111–5120. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Vijayasarathy, C.; Zeng, Y.; Marangoni, D.; Bush, R.A.; Wu, Z.; Sieving, P.A. NADPH Oxidase Contributes to Photoreceptor Degeneration in Constitutively Active RAC1 Mice. Investig. Ophthalmol. Vis. Sci. 2016, 57, 2864–2875. [Google Scholar] [CrossRef] [Green Version]
- Brown, G.C.; Borutaite, V. Nitric oxide, mitochondria, and cell death. IUBMB Life 2001, 52, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Durango, R.; Fernandez-Martinez, A.; Garcia-Feijoo, J.; Castillo, A.; de la Casa, J.M.; Garcia-Bueno, B.; Perez-Nievas, B.G.; Fernandez-Cruz, A.; Leza, J.C. Expression of nitrotyrosine and oxidative consequences in the trabecular meshwork of patients with primary open-angle glaucoma. Invest. Ophthalmol. Vis. Sci. 2008, 49, 2506–2511. [Google Scholar] [CrossRef] [Green Version]
- Ghanem, A.A.; Elewa, A.M.; Arafa, L.F. Endothelin-1 and nitric oxide levels in patients with glaucoma. Ophthalmic Res. 2011, 46, 98–102. [Google Scholar] [CrossRef]
- Noske, W.; Hensen, J.; Wiederholt, M. Endothelin-like immunoreactivity in aqueous humor of patients with primary open-angle glaucoma and cataract. Graefe’s Arch. Clin. Exp. Ophthalmol. 1997, 235, 551–552. [Google Scholar] [CrossRef] [PubMed]
- Choritz, L.; Machert, M.; Thieme, H. Correlation of endothelin-1 concentration in aqueous humor with intraocular pressure in primary open angle and pseudoexfoliation glaucoma. Investig. Ophthalmol. Vis. Sci. 2012, 53, 7336–7342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wareham, L.K.; Calkins, D.J. The Neurovascular Unit in Glaucomatous Neurodegeneration. Front. Cell Dev. Biol. 2020, 8, 452. [Google Scholar] [CrossRef] [PubMed]
- Jansen, E.H.; Ruskovska, T. Comparative Analysis of Serum (Anti)oxidative Status Parsmall a, Cyrillicmeters in Healthy Persons. Int. J. Mol. Sci. 2013, 14, 6106–6115. [Google Scholar] [CrossRef] [Green Version]
- Abu-Amero, K.K.; Kondkar, A.A.; Mousa, A.; Osman, E.A.; Al-Obeidan, S.A. Decreased total antioxidants in patients with primary open angle glaucoma. Curr. Eye Res. 2013, 38, 959–964. [Google Scholar] [CrossRef]
- Ruskovska, T.; Jansen, E.H.; Antarorov, R. Evaluation of assays for measurement of serum (anti)oxidants in hemodialysis patients. BioMed Res. Int. 2014, 2014, 843157. [Google Scholar] [CrossRef] [Green Version]
- Tanito, M.; Kaidzu, S.; Takai, Y.; Ohira, A. Association between systemic oxidative stress and visual field damage in open-angle glaucoma. Sci. Rep. 2016, 6, 25792. [Google Scholar] [CrossRef] [Green Version]
- Kilic, R.; Bayraktar, A.C.; Bayraktar, S.; Kurt, A.; Kavutcu, M. Evaluation of Serum Superoxide Dismutase Activity, Malondialdehyde, and Zinc and Copper Levels in Patients With Keratoconus. Cornea 2016, 35, 1512–1515. [Google Scholar] [CrossRef]
- Nucci, C.; Di Pierro, D.; Varesi, C.; Ciuffoletti, E.; Russo, R.; Gentile, R.; Cedrone, C.; Pinazo Duran, M.D.; Coletta, M.; Mancino, R. Increased malondialdehyde concentration and reduced total antioxidant capacity in aqueous humor and blood samples from patients with glaucoma. Mol. Vis. 2013, 19, 1841–1846. [Google Scholar]
- Mousa, A.; Kondkar, A.A.; Al-Obeidan, S.A.; Azad, T.A.; Sultan, T.; Osman, E.; Abu-Amero, K.K. Association of total antioxidants level with glaucoma type and severity. Saudi Med. J. 2015, 36, 671–677. [Google Scholar] [CrossRef]
- Bosco, A.; Steele, M.R.; Vetter, M.L. Early microglia activation in a mouse model of chronic glaucoma. J. Comp. Neurol. 2011, 519, 599–620. [Google Scholar] [CrossRef] [Green Version]
- Tribble, J.R.; Harder, J.M.; Williams, P.A.; John, S.W.M. Ocular hypertension suppresses homeostatic gene expression in optic nerve head microglia of DBA/2 J mice. Mol. Brain 2020, 13, 81. [Google Scholar] [CrossRef]
- Tezel, G.; Yang, J.; Wax, M.B. Heat shock proteins, immunity and glaucoma. Brain Res. Bull. 2004, 62, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G. TNF-alpha signaling in glaucomatous neurodegeneration. Prog. Brain Res. 2008, 173, 409–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Cho, K.S.; Vu, T.H.K.; Shen, C.H.; Kaur, M.; Chen, G.; Mathew, R.; McHam, M.L.; Fazelat, A.; Lashkari, K.; et al. Commensal microflora-induced T cell responses mediate progressive neurodegeneration in glaucoma. Nat. Commun. 2018, 9, 3209. [Google Scholar] [CrossRef] [Green Version]
- Tsai, T.; Grotegut, P.; Reinehr, S.; Joachim, S.C. Role of Heat Shock Proteins in Glaucoma. Int. J. Mol. Sci. 2019, 20, 5160. [Google Scholar] [CrossRef] [Green Version]
- Soto, I.; Howell, G.R. The complex role of neuroinflammation in glaucoma. Cold Spring Harb. Perspect. Med. 2014, 4, a017269. [Google Scholar] [CrossRef]
- Chen, F.; Pandey, D.; Chadli, A.; Catravas, J.D.; Chen, T.; Fulton, D.J. Hsp90 regulates NADPH oxidase activity and is necessary for superoxide but not hydrogen peroxide production. Antioxid. Redox Signal. 2011, 14, 2107–2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.; Cho, K.S.; Thee, E.F.; Jager, M.J.; Chen, D.F. Neuroinflammation and microglia in glaucoma: Time for a paradigm shift. J. Neurosci. Res. 2019, 97, 70–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, A.I.; de Hoz, R.; Salobrar-Garcia, E.; Salazar, J.J.; Rojas, B.; Ajoy, D.; Lopez-Cuenca, I.; Rojas, P.; Trivino, A.; Ramirez, J.M. The Role of Microglia in Retinal Neurodegeneration: Alzheimer’s Disease, Parkinson, and Glaucoma. Front. Aging Neurosci. 2017, 9, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, Z.; Guo, Y.; Shrestha, M.; Sun, D.; Gregory-Ksander, M.; Jakobs, T.C. Microglia depletion exacerbates retinal ganglion cell loss in a mouse model of glaucoma. Exp. Eye Res. 2022, 225, 109273. [Google Scholar] [CrossRef]
- Tanaka-Gonome, T.; Xie, Y.; Yamauchi, K.; Maeda-Monai, N.; Tanabu, R.; Kudo, T.; Nakazawa, M. The protective effect of astaxanthin on the ganglion cell complex in glutamate/aspartate transporter deficient mice, a model of normal tension glaucoma, analyzed by spectral domain-optical coherence tomography. Biochem. Biophys. Rep. 2020, 23, 100777. [Google Scholar] [CrossRef]
- Angel Aillegas, N.; Tartara, L.I.; Caballero, G.; Campana, V.; Allemandi, D.A.; Palma, S.D. Antioxidant status in rabbit aqueous humor after instillation of ascorbyl laurate-based nanostructures. Pharmacol. Rep. 2019, 71, 794–797. [Google Scholar] [CrossRef]
- Harris, A.; Gross, J.; Moore, N.; Do, T.; Huang, A.; Gama, W.; Siesky, B. The effects of antioxidants on ocular blood flow in patients with glaucoma. Acta Ophthalmol. 2018, 96, e237–e241. [Google Scholar] [CrossRef] [Green Version]
- Ozates, S.; Elgin, K.U.; Yilmaz, N.S.; Demirel, O.O.; Sen, E.; Yilmazbas, P. Evaluation of oxidative stress in pseudo-exfoliative glaucoma patients treated with and without topical coenzyme Q10 and vitamin E. Eur. J. Ophthalmol. 2019, 29, 196–201. [Google Scholar] [CrossRef]
- Romeo Villadoniga, S.; Rodriguez Garcia, E.; Sagastagoia Epelde, O.; Alvarez Diaz, M.D.; Domingo Pedrol, J.C. Effects of Oral Supplementation with Docosahexaenoic Acid (DHA) plus Antioxidants in Pseudoexfoliative Glaucoma: A 6-Month Open-Label Randomized Trial. J. Ophthalmol. 2018, 2018, 8259371. [Google Scholar] [CrossRef] [Green Version]
- Sinclair, D.A.; LaPlante, M.D. Lifespan: Why We Age—And Why We Don’t Have To; Simon and Schuster: New York, NY, USA, 2019. [Google Scholar]
- Barzilai, N. Age Later: Health Span, Life Span, and the New Science of Longevity; St. Martin’s Press: New York, NY, USA, 2020. [Google Scholar]
- Mitnitski, A.B.; Graham, J.E.; Mogilner, A.J.; Rockwood, K. Frailty, fitness and late-life mortality in relation to chronological and biological age. BMC Geriatr. 2002, 2, 1. [Google Scholar] [CrossRef] [Green Version]
- Schultz, M.B.; Kane, A.E.; Mitchell, S.J.; MacArthur, M.R.; Warner, E.; Vogel, D.S.; Mitchell, J.R.; Howlett, S.E.; Bonkowski, M.S.; Sinclair, D.A. Age and life expectancy clocks based on machine learning analysis of mouse frailty. Nat. Commun. 2020, 11, 4618. [Google Scholar] [CrossRef]
- Rowe, J.W.; Kahn, R.L. Successful aging. Gerontologist 1997, 37, 433–440. [Google Scholar] [CrossRef]
- Sinha, M.; Jang, Y.C.; Oh, J.; Khong, D.; Wu, E.Y.; Manohar, R.; Miller, C.; Regalado, S.G.; Loffredo, F.S.; Pancoast, J.R. Restoring systemic GDF11 levels reverses age-related dysfunction in mouse skeletal muscle. Science 2014, 344, 649–652. [Google Scholar] [CrossRef] [Green Version]
- Katsimpardi, L.; Litterman, N.K.; Schein, P.A.; Miller, C.M.; Loffredo, F.S.; Wojtkiewicz, G.R.; Chen, J.W.; Lee, R.T.; Wagers, A.J.; Rubin, L.L. Vascular and neurogenic rejuvenation of the aging mouse brain by young systemic factors. Science 2014, 344, 630–634. [Google Scholar] [CrossRef] [Green Version]
- Mattson, M.P. Hormesis defined. Ageing Res. Rev. 2008, 7, 1–7. [Google Scholar] [CrossRef]
- Martin-Montalvo, A.; Mercken, E.M.; Mitchell, S.J.; Palacios, H.H.; Mote, P.L.; Scheibye-Knudsen, M.; Gomes, A.P.; Ward, T.M.; Minor, R.K.; Blouin, M.J.; et al. Metformin improves healthspan and lifespan in mice. Nat. Commun. 2013, 4, 2192. [Google Scholar] [CrossRef] [Green Version]
- Bordone, L.; Guarente, L. Calorie restriction, SIRT1 and metabolism: Understanding longevity. Nat. Rev. Mol. Cell Biol. 2005, 6, 298–305. [Google Scholar] [CrossRef]
- Garay, R.P. Investigational drugs and nutrients for human longevity. Recent clinical trials registered in ClinicalTrials.gov and clinicaltrialsregister.eu. Expert Opin. Investig. Drugs 2021, 30, 749–758. [Google Scholar] [CrossRef]
- Bailey, C.J.; Turner, R.C. Metformin. N. Engl. J. Med. 1996, 334, 574–579. [Google Scholar] [CrossRef]
- De Haes, W.; Frooninckx, L.; Van Assche, R.; Smolders, A.; Depuydt, G.; Billen, J.; Braeckman, B.P.; Schoofs, L.; Temmerman, L. Metformin promotes lifespan through mitohormesis via the peroxiredoxin PRDX-2. Proc. Natl. Acad. Sci. USA 2014, 111, E2501–E2509. [Google Scholar] [CrossRef] [Green Version]
- Hurley, D.J.; Irnaten, M.; O’Brien, C. Metformin and Glaucoma—Review of Anti-Fibrotic Processes and Bioenergetics. Cells 2021, 10, 2131. [Google Scholar] [CrossRef]
- Brown, E.E.; Lewin, A.S.; Ash, J.D. Mitochondria: Potential targets for protection in age-related macular degeneration. Retin. Degener. Dis. 2018, 1074, 11–17. [Google Scholar]
- Amin, S.V.; Khanna, S.; Parvar, S.P.; Shaw, L.T.; Dao, D.; Hariprasad, S.M.; Skondra, D. Metformin and retinal diseases in preclinical and clinical studies: Insights and review of literature. Exp. Biol. Med. 2022, 247, 317–329. [Google Scholar] [CrossRef]
- Romdhoniyyah, D.F.; Harding, S.P.; Cheyne, C.P.; Beare, N.A. Metformin, a potential role in age-related macular degeneration: A systematic review and meta-analysis. Ophthalmol. Ther. 2021, 10, 245–260. [Google Scholar] [CrossRef]
- Onyango, P.; Celic, I.; McCaffery, J.M.; Boeke, J.D.; Feinberg, A.P. SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proc. Natl. Acad. Sci. USA 2002, 99, 13653–13658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Kim, Y.; Liu, T.; Hwang, Y.J.; Hyeon, S.J.; Im, H.; Lee, K.; Alvarez, V.E.; McKee, A.C.; Um, S.-J.; et al. SIRT3 deregulation is linked to mitochondrial dysfunction in Alzheimer’s disease. Aging Cell 2018, 17, e12679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, A.; Wang, J.; Ghena, N.; Zhao, Q.; Perone, I.; King, T.M.; Veech, R.L.; Gorospe, M.; Wan, R.; Mattson, M.P. SIRT3 Haploinsufficiency Aggravates Loss of GABAergic Interneurons and Neuronal Network Hyperexcitability in an Alzheimer’s Disease Model. J. Neurosci. 2020, 40, 694–709. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Bhawal, R.; Xu, H.; Chen, H.; Anderson, E.T.; Haroutunian, V.; Cross, A.C.; Zhang, S.; Gibson, G.E. The human brain acetylome reveals that decreased acetylation of mitochondrial proteins associates with Alzheimer’s disease. J. Neurochem. 2021, 158, 282–296. [Google Scholar] [CrossRef]
- Tyagi, A.; Nguyen, C.U.; Chong, T.; Michel, C.R.; Fritz, K.S.; Reisdorph, N.; Knaub, L.; Reusch, J.E.B.; Pugazhenthi, S. SIRT3 deficiency-induced mitochondrial dysfunction and inflammasome formation in the brain. Sci. Rep. 2018, 8, 17547. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Sun, Y.-M.; Huang, H.; Chen, C.; Wan, J.; Ma, L.-H.; Sun, Y.-Y.; Miao, H.-H.; Wu, Y.-Q. Sirtuin 3 protects against anesthesia/surgery-induced cognitive decline in aged mice by suppressing hippocampal neuroinflammation. J. Neuroinflamm. 2021, 18, 41. [Google Scholar] [CrossRef]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [CrossRef] [Green Version]
- Forman, H.J.; Fukuto, J.M.; Torres, M. Redox signaling: Thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. Am. J. Physiol. Cell Physiol. 2004, 287, C246–C256. [Google Scholar] [CrossRef]
- Torres, M. Mitogen-activated protein kinase pathways in redox signaling. Front. Biosci. 2003, 8, d369–d391. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Barrios, A.; Alvarez, L.; Garcia, M.; Artime, E.; Pereiro, R.; Gonzalez-Iglesias, H. Antioxidant Defenses in the Human Eye: A Focus on Metallothioneins. Antioxidants 2021, 10, 89. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Redox homeostasis in the eye. A homeostatic balance of antioxidants and reactive oxygen species (ROS) is required for the healthy functioning of ocular tissues. An imbalance of antioxidants (AOX) and ROS through either the depletion of antioxidants or excessive ROS accumulation will result in oxidative stress and drive subsequent disease progression. Antioxidants are categorized as either enzymatic or non-enzymatic. ROS can be derived from either endogenous or exogenous sources. Examples of heavy metals include lead (Pb), arsenic (As), and mercury (Hg).
Figure 1.
Redox homeostasis in the eye. A homeostatic balance of antioxidants and reactive oxygen species (ROS) is required for the healthy functioning of ocular tissues. An imbalance of antioxidants (AOX) and ROS through either the depletion of antioxidants or excessive ROS accumulation will result in oxidative stress and drive subsequent disease progression. Antioxidants are categorized as either enzymatic or non-enzymatic. ROS can be derived from either endogenous or exogenous sources. Examples of heavy metals include lead (Pb), arsenic (As), and mercury (Hg).

Figure 2.
Factors driving oxidative damage during age-related macular degeneration (AMD) and the need for antioxidant defense systems to combat the pathophysiology of the disease.
Figure 2.
Factors driving oxidative damage during age-related macular degeneration (AMD) and the need for antioxidant defense systems to combat the pathophysiology of the disease.

Figure 3.
Diabetes-induced hyperglycemia disrupts the redox balance, leading to oxidative stress in the retina. Under normal physiological conditions, there is a balance in the production of reactive oxygen species (ROS)/reactive nitrogen species (RNS) and the antioxidant defense system. Diabetes-induced hyperglycemia promotes ROS/RNS generation while also suppressing the retinal antioxidant response, creating the imbalance known as oxidative stress. The AGE pathway, the polyol pathway, the hexosamine biosynthetic pathway (HBP), and the protein kinase C pathway are all sensitive to this disruption and play an important role in the downstream effects of hyperglycemia-induced retinal damage.
Figure 3.
Diabetes-induced hyperglycemia disrupts the redox balance, leading to oxidative stress in the retina. Under normal physiological conditions, there is a balance in the production of reactive oxygen species (ROS)/reactive nitrogen species (RNS) and the antioxidant defense system. Diabetes-induced hyperglycemia promotes ROS/RNS generation while also suppressing the retinal antioxidant response, creating the imbalance known as oxidative stress. The AGE pathway, the polyol pathway, the hexosamine biosynthetic pathway (HBP), and the protein kinase C pathway are all sensitive to this disruption and play an important role in the downstream effects of hyperglycemia-induced retinal damage.


{kind=link}
{kind=link}
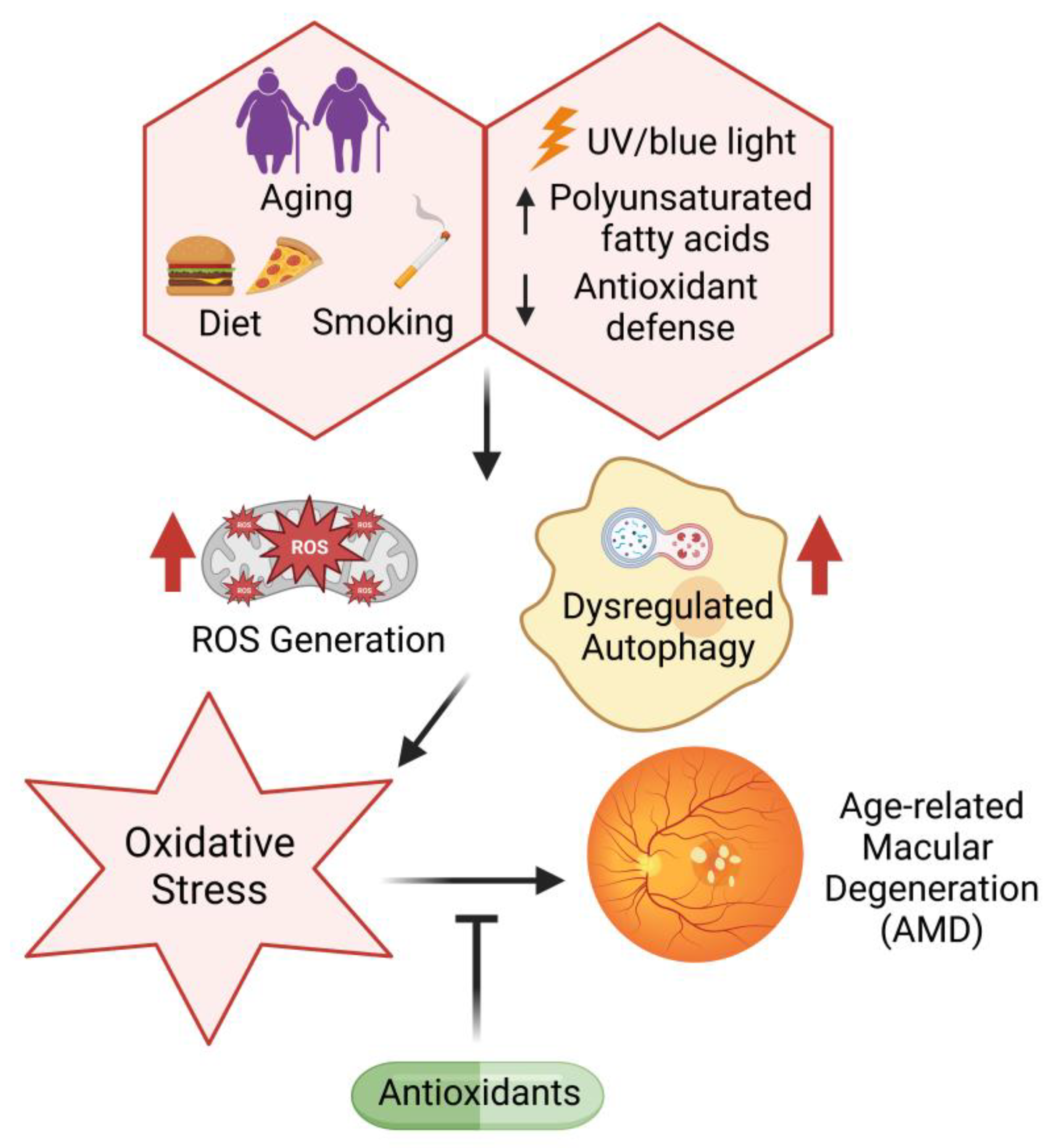{kind=link}
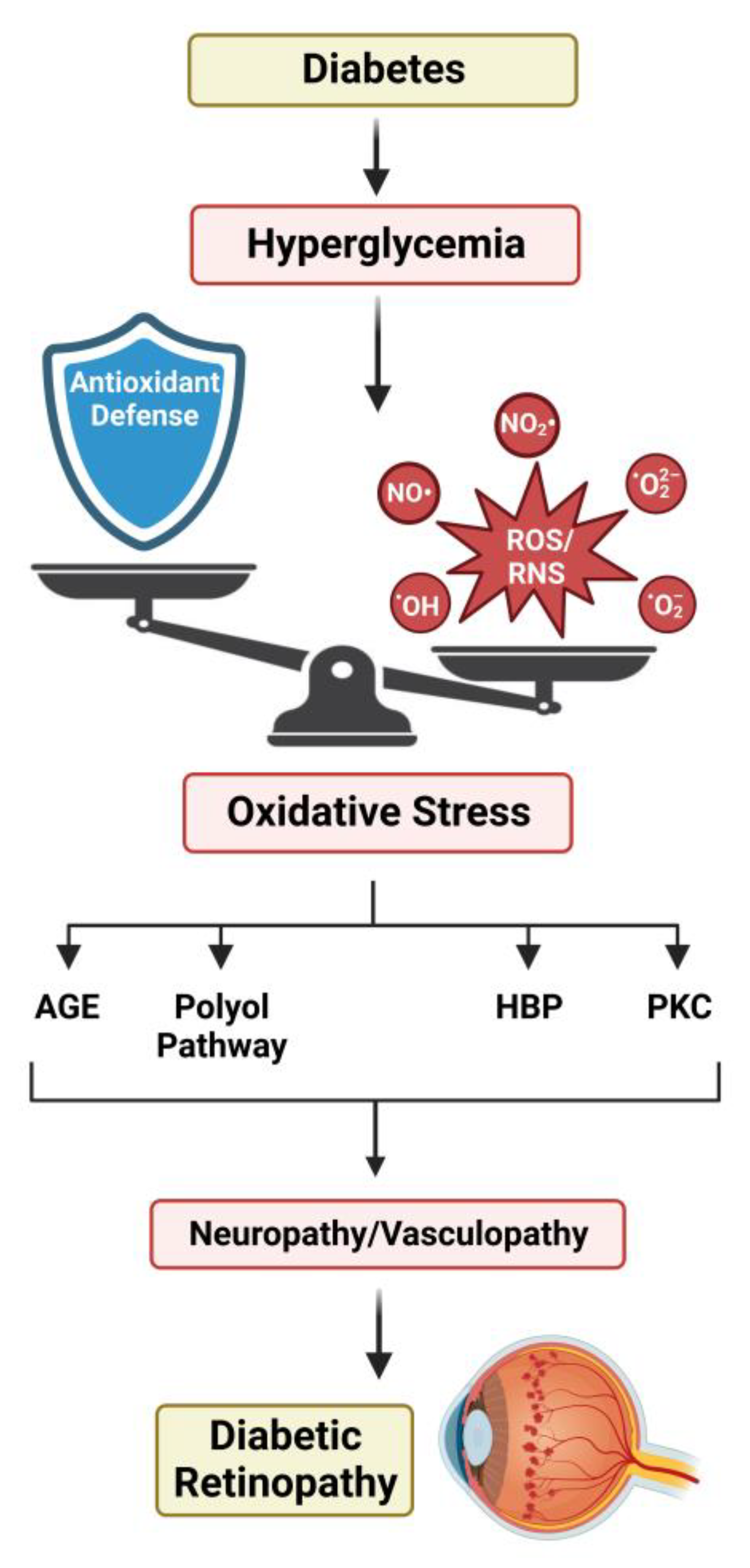{kind=link}
Table 1.
Summary of the major chemical species associated with oxidative stress in the eye.
| Chemical Species | Source(s) | Downstream Reaction(s) | Ref. |
|---|---|---|---|
| Superoxide (O2·−) | The reaction of O2 with enzymes in the electron transport chain in the mitochondria generated via a single-electron transfer; enzymatic and non-enzymatic biosynthetic pathways; produced by neutrophils | Reacts with O2·− and H2O to generate H2O2 and O2 | [13,14,15] |
| Hydroxyl radical (·OH) | The reaction of H2O2 with iron or copper (Fenton reaction); may also be generated as a byproduct of the exposure of water molecules to ionizing radiation | Reacts with deoxyguanosine residues of DNA to form 8-hydroxy-2-deoxyguanosine; also reacts with deoxycytidine and deoxyadenosine, among others | [16,17] |
| Hydrogen peroxide (H2O2) | The reaction of O2·− molecules mediated via superoxide dismutase and non-enzymatically; may also be generated as a byproduct of normal catalytic oxidative processes mediated via oxidases | May be converted by myeloperoxidase or other enzymes containing Fe2+ or react with UV light to form hydroxyl radical (OH·); involved in downstream signaling pathways, such as platelet-derived growth factor signaling | [18,19] |
| Malondialdehyde (MDA) (CH2(CHO)2) | Produced by lipid peroxidation of polyunsaturated fatty acids | Reacts with deoxyguanosine of DNA to form 8-hydroxy-2-deoxyguanosine; also reacts with deoxyadenosine residues; may also react with lysine residues on proteins to form secondary oxidation products | [20,21] |
| 4-Hydroxynonenal (4-HNE) (CH3(CH2)4CH(OH)CH=CH(CHO)) | Produced by lipid peroxidation of polyunsaturated fatty acids or linoleic or arachidonic side chains | Reacts with lysine on proteins to form carbonylated side chains, increasing the hydrophobicity of modified proteins; also involved in downstream signaling pathways, including activation of glutamate-cysteine ligase expression | [22,23] |
Table 2.
Summary of key findings associating oxidative stress with pathologies affecting the anterior and posterior segments of the eye (Abbreviations: electron transport chain (ETC); reduced glutathione (GSH); 4-hydroxynonenal (4-HNE); malondialdehyde (MDA); nuclear factor-erythroid 2 related factor 2 (NRF2); 8-hydroxy-2-deoxyguanosine (8-OHdG); retinal ganglion cells (RGCs); retinal pigment epithelium (RPE); superoxide dismutase (SOD); tricarboxylic acid (TCA)).
Table 2.
Summary of key findings associating oxidative stress with pathologies affecting the anterior and posterior segments of the eye (Abbreviations: electron transport chain (ETC); reduced glutathione (GSH); 4-hydroxynonenal (4-HNE); malondialdehyde (MDA); nuclear factor-erythroid 2 related factor 2 (NRF2); 8-hydroxy-2-deoxyguanosine (8-OHdG); retinal ganglion cells (RGCs); retinal pigment epithelium (RPE); superoxide dismutase (SOD); tricarboxylic acid (TCA)).
| Condition | Clinical Manifestation(s) | Identified Markers of Oxidative Stress | Ref. |
|---|---|---|---|
| Dry eye disease | Instability of the lipid layer, decreased tear secretion, ocular irritation | Elevated 8-OHdG, 4-HNE, and MDA and higher immune infiltration in dry eye animal models; elevated 4-HNE and hexanoyl-lysine in the conjunctiva of patients with dry eye and Sjögren’s syndrome | [42,43,45] |
| Keratoconus | Thinning of the corneal stroma leading to bulging of the central cornea | Elevated lipid peroxidation, MDA, and proinflammatory cytokines in tears of patients with keratoconus; increased lactate production and altered glycolytic and TCA cycle metabolite levels in corneal fibroblasts; decreased NRF2 expression | [64,65,66,67] |
| Cataract | Opacification of the lens | Oxidation, deamidation, and other chemical modifications of crystallin proteins, leading to protein aggregation and precipitation; decreased GSH in lens epithelial cells | [117,121,138] |
| Age-related macular degeneration | Drusen formation and photoreceptor degeneration; choroidal neovascularization, hemorrhage, and retinal fibrosis | Elevated MDA and 8-OHdG; decreased SOD in RPE cells; elevated carboxyethylpyrrole in drusen | [221,225,233] |
| Proliferative vitreoretinopathy | Formation of fibrotic membranes on the retinal surface | Reduced SOD and catalase in vitreous; decreased GSH in vitreous and blood | [283,284] |
| Diabetic retinopathy | Vascular abnormalities in the retina; microaneurysms, hemorrhaging, and angiogenesis | Increased ROS production by ETC with hyperglycemia; decreased SOD, catalase, and GSH production | [321,322,329] |
| Glaucoma | Loss of RGCs in the retina | Reduced total reactive antioxidant potential and increased MDA in the aqueous humor and blood | [381,382,424] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Shu, D.Y.; Chaudhary, S.; Cho, K.-S.; Lennikov, A.; Miller, W.P.; Thorn, D.C.; Yang, M.; McKay, T.B. Role of Oxidative Stress in Ocular Diseases: A Balancing Act. Metabolites 2023, 13, 187. https://doi.org/10.3390/metabo13020187
AMA Style
Shu DY, Chaudhary S, Cho K-S, Lennikov A, Miller WP, Thorn DC, Yang M, McKay TB. Role of Oxidative Stress in Ocular Diseases: A Balancing Act. Metabolites. 2023; 13(2):187. https://doi.org/10.3390/metabo13020187
Chicago/Turabian StyleShu, Daisy Y., Suman Chaudhary, Kin-Sang Cho, Anton Lennikov, William P. Miller, David C. Thorn, Menglu Yang, and Tina B. McKay. 2023. "Role of Oxidative Stress in Ocular Diseases: A Balancing Act" Metabolites 13, no. 2: 187. https://doi.org/10.3390/metabo13020187
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.