
Bruch’s Membrane: A Key Consideration with Complement-Based Therapies for Age-Related Macular Degeneration

Abstract
:
1. Introduction
2. The Structure and Function of BrM
- The central elastic layer (CEL) is about 0.8 μm thick, discontinuous in the macular region [27], and contains elastin fibres that are contiguous with the ICL and outer collagenous layer (OCL). The CEL is important for biomechanical properties, antiangiogenic barrier functions, and choroidal contractility [28].
- The choroidal endothelial cell (CEC) basement membrane is about 0.07 μm thick and contains collagen IV, α1, α2, V, and VI, heparan sulphate, laminin, endostatin, and chondroitin sulphate [15,19,20,33,34]. It is discontinuous due to the presence of choroidal inter-capillary pillars between CC lumens [28].
3. Ageing Processes in BrM
3.1. Anatomical Changes
3.2. Biomechanical Changes
3.3. Permeability Changes
4. Overview of AMD
4.1. Genetic Risk Factors
4.2. Environmental Risk Factors

5. Non-Invasive Imaging of BrM
5.1. In Vivo Imaging of BrM in AMD
5.2. In Vivo Imaging of BrM in Inherited Retinal Degenerations (IRDs)
6. Overview of Complement
7. Complement Dysfunction in AMD
7.1. The Role of Complement in Choroidal Homeostasis
7.2. The Role of Complement in RPE Function
7.3. Impact of Complement Overactivation on RPE Function and Viability
7.4. Complement Dysfunction in Macular Neovascularisation
7.5. Immune Cell Regulation
8. Local vs. Systemic Complement Production in AMD
9. Complement as a Therapeutic Target
9.1. Systemic Therapies
9.2. Intravitreal Therapies
9.3. Subretinal Therapies
9.4. Suprachoroidal Therapies
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.Q.; Welchowski, T.; Schmid, M.; Mauschitz, M.M.; Holz, F.G.; Finger, R.P. Prevalence and incidence of age-related macular degeneration in Europe: A systematic review and meta-analysis. Br. J. Ophthalmol. 2020, 104, 1077–1084. [Google Scholar] [CrossRef]
- Minassian, D.; Reidy, A. Future Sight Loss UK (2): An Epidemiological and Economic Model for Sight Loss in the Decade 2010–2020. 2009. Available online: https://media.rnib.org.uk/documents/FSUK_Report_2_0.doc (accessed on 2 April 2023).
- Chakravarthy, U.; Bailey, C.C.; Johnston, R.L.; McKibbin, M.; Khan, R.S.; Mahmood, S.; Downey, L.; Dhingra, N.; Brand, C.; Brittain, C.J.; et al. Characterizing Disease Burden and Progression of Geographic Atrophy Secondary to Age-Related Macular Degeneration. Ophthalmology 2018, 125, 842–849. [Google Scholar] [CrossRef] [Green Version]
- Rofagha, S.; Bhisitkul, R.B.; Boyer, D.S.; Sadda, S.R.; Zhang, K.; Group, S.-U.S. Seven-year outcomes in ranibizumab-treated patients in ANCHOR, MARINA, and HORIZON: A multicenter cohort study (SEVEN-UP). Ophthalmology 2013, 120, 2292–2299. [Google Scholar] [CrossRef]
- Guillonneau, X.; Eandi, C.M.; Paques, M.; Sahel, J.A.; Sapieha, P.; Sennlaub, F. On phagocytes and macular degeneration. Prog. Retin. Eye Res. 2017, 61, 98–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzoumas, N.; Hallam, D.; Harris, C.L.; Lako, M.; Kavanagh, D.; Steel, D.H.W. Revisiting the role of factor H in age-related macular degeneration: Insights from complement-mediated renal disease and rare genetic variants. Surv. Ophthalmol. 2021, 66, 378–401. [Google Scholar] [CrossRef] [PubMed]
- Pauly, D.; Agarwal, D.; Dana, N.; Schafer, N.; Biber, J.; Wunderlich, K.A.; Jabri, Y.; Straub, T.; Zhang, N.R.; Gautam, A.K.; et al. Cell-Type-Specific Complement Expression in the Healthy and Diseased Retina. Cell. Rep. 2019, 29, 2835–2848.E4. [Google Scholar] [CrossRef]
- Lakkaraju, A.; Umapathy, A.; Tan, L.X.; Daniele, L.; Philp, N.J.; Boesze-Battaglia, K.; Williams, D.S. The cell biology of the retinal pigment epithelium. Prog. Retin. Eye Res. 2020, 78, 100846. [Google Scholar] [CrossRef] [PubMed]
- Nickla, D.L.; Wallman, J. The multifunctional choroid. Prog. Retin. Eye Res. 2010, 29, 144–168. [Google Scholar] [CrossRef] [Green Version]
- Edwards, M.; Lutty, G.A. Bruch’s Membrane and the Choroid in Age-Related Macular Degeneration. Adv. Exp. Med. Biol. 2021, 1256, 89–119. [Google Scholar] [CrossRef]
- Hogan, M.J.; Alvarado, A.J.; Weddell, J.E. Histology of the Human. Eye: An. Atlas and Textbook; Saunders: Philadelphia, PA, USA, 1971. [Google Scholar]
- Guymer, R.; Luthert, P.; Bird, A. Changes in Bruch’s membrane and related structures with age. Prog. Retin. Eye Res. 1999, 18, 59–90. [Google Scholar] [CrossRef] [PubMed]
- Booij, J.C.; Baas, D.C.; Beisekeeva, J.; Gorgels, T.G.; Bergen, A.A. The dynamic nature of Bruch’s membrane. Prog. Retin. Eye Res. 2010, 29, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Miyamura, N.; Ninomiya, Y.; Handa, J.T. Distribution of the collagen IV isoforms in human Bruch’s membrane. Br. J. Ophthalmol. 2003, 87, 212–215. [Google Scholar] [CrossRef] [Green Version]
- Aisenbrey, S.; Zhang, M.; Bacher, D.; Yee, J.; Brunken, W.J.; Hunter, D.D. Retinal pigment epithelial cells synthesize laminins, including laminin 5, and adhere to them through alpha3- and alpha6-containing integrins. Investig. Ophthalmol. Vis. Sci. 2006, 47, 5537–5544. [Google Scholar] [CrossRef] [PubMed]
- Call, T.W.; Hollyfield, J.G. Sulfated proteoglycans in Bruch’s membrane of the human eye: Localization and characterization using cupromeronic blue. Exp. Eye Res. 1990, 51, 451–462. [Google Scholar] [CrossRef]
- Beattie, J.R.; Pawlak, A.M.; Boulton, M.E.; Zhang, J.; Monnier, V.M.; McGarvey, J.J.; Stitt, A.W. Multiplex analysis of age-related protein and lipid modifications in human Bruch’s membrane. FASEB J. 2010, 24, 4816–4824. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Frank, R.N.; Zhang, N.L.; Turczyn, T.J. Ultrastructural localization of extracellular matrix components in human retinal vessels and Bruch’s membrane. Arch. Ophthalmol. 1990, 108, 421–429. [Google Scholar] [CrossRef]
- Lin, W.L.; Essner, E.; McCarthy, K.J.; Couchman, J.R. Ultrastructural immunocytochemical localization of chondroitin sulfate proteoglycan in Bruch’s membrane of the rat. Investig. Ophthalmol. Vis. Sci. 1992, 33, 2072–2075. [Google Scholar]
- Marmor, M.F.; Wolfensberger, T.J. The Retinal Pigment Epithelium; Oxford University Press: New York, NY, USA, 1998. [Google Scholar]
- Ruberti, J.W.; Curcio, C.A.; Millican, C.L.; Menco, B.P.; Huang, J.D.; Johnson, M. Quick-freeze/deep-etch visualization of age-related lipid accumulation in Bruch’s membrane. Investig. Ophthalmol. Vis. Sci. 2003, 44, 1753–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hageman, G.S.; Mullins, R.F.; Russell, S.R.; Johnson, L.V.; Anderson, D.H. Vitronectin is a constituent of ocular drusen and the vitronectin gene is expressed in human retinal pigmented epithelial cells. FASEB J. 1999, 13, 477–484. [Google Scholar] [CrossRef]
- Anderson, D.H.; Ozaki, S.; Nealon, M.; Neitz, J.; Mullins, R.F.; Hageman, G.S.; Johnson, L.V. Local cellular sources of apolipoprotein E in the human retina and retinal pigmented epithelium: Implications for the process of drusen formation. Am. J. Ophthalmol. 2001, 131, 767–781. [Google Scholar] [CrossRef] [PubMed]
- Tezel, T.H.; Geng, L.; Lato, E.B.; Schaal, S.; Liu, Y.; Dean, D.; Klein, J.B.; Kaplan, H.J. Synthesis and secretion of hemoglobin by retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1911–1919. [Google Scholar] [CrossRef]
- Sakaguchi, H.; Miyagi, M.; Shadrach, K.G.; Rayborn, M.E.; Crabb, J.W.; Hollyfield, J.G. Clusterin is present in drusen in age-related macular degeneration. Exp. Eye Res. 2002, 74, 547–549. [Google Scholar] [CrossRef]
- Chong, N.H.; Keonin, J.; Luthert, P.J.; Frennesson, C.I.; Weingeist, D.M.; Wolf, R.L.; Mullins, R.F.; Hageman, G.S. Decreased thickness and integrity of the macular elastic layer of Bruch’s membrane correspond to the distribution of lesions associated with age-related macular degeneration. Am. J. Pathol. 2005, 166, 241–251. [Google Scholar] [CrossRef]
- Johnson, M.; Curcio, C.A. Structure, Function, and Pathology of Bruch’s Membrane; Elsevier: Amsterdam, The Netherlands, 2012; Volume 1. [Google Scholar]
- Newsome, D.A.; Huh, W.; Green, W.R. Bruch’s membrane age-related changes vary by region. Curr. Eye Res. 1987, 6, 1211–1221. [Google Scholar] [CrossRef]
- Huang, J.D.; Presley, J.B.; Chimento, M.F.; Curcio, C.A.; Johnson, M. Age-related changes in human macular Bruch’s membrane as seen by quick-freeze/deep-etch. Exp. Eye Res. 2007, 85, 202–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Li, C.M.; Rudolf, M.; Belyaeva, O.V.; Chung, B.H.; Messinger, J.D.; Kedishvili, N.Y.; Curcio, C.A. Lipoprotein particles of intraocular origin in human Bruch membrane: An unusual lipid profile. Investig. Ophthalmol. Vis. Sci. 2009, 50, 870–877. [Google Scholar] [CrossRef]
- Mullins, R.F.; Olvera, M.A.; Clark, A.F.; Stone, E.M. Fibulin-5 distribution in human eyes: Relevance to age-related macular degeneration. Exp. Eye Res. 2007, 84, 378–380. [Google Scholar] [CrossRef] [Green Version]
- Bhutto, I.A.; Kim, S.Y.; McLeod, D.S.; Merges, C.; Fukai, N.; Olsen, B.R.; Lutty, G.A. Localization of collagen XVIII and the endostatin portion of collagen XVIII in aged human control eyes and eyes with age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2004, 45, 1544–1552. [Google Scholar] [CrossRef] [Green Version]
- Yamada, Y.; Tian, J.; Yang, Y.; Cutler, R.G.; Wu, T.; Telljohann, R.S.; Mattson, M.P.; Handa, J.T. Oxidized low density lipoproteins induce a pathologic response by retinal pigmented epithelial cells. J. Neurochem. 2008, 105, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; McHarg, S.; Tilakaratna, V.; Brace, N.; Bishop, P.N. Bruch’s Membrane Compartmentalizes Complement Regulation in the Eye with Implications for Therapeutic Design in Age-Related Macular Degeneration. Front. Immunol. 2017, 8, 1778. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.J.; Schmidt, C.Q.; White, A.M.; Hakobyan, S.; Morgan, B.P.; Bishop, P.N. Identification of factor H-like protein 1 as the predominant complement regulator in Bruch’s membrane: Implications for age-related macular degeneration. J. Immunol. 2014, 193, 4962–4970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langford-Smith, A.; Day, A.J.; Bishop, P.N.; Clark, S.J. Complementing the Sugar Code: Role of GAGs and Sialic Acid in Complement Regulation. Front. Immunol. 2015, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Bishop, P.N.; Day, A.J. The proteoglycan glycomatrix: A sugar microenvironment essential for complement regulation. Front. Immunol. 2013, 4, 412. [Google Scholar] [CrossRef] [Green Version]
- Tsiftsoglou, S.A.; Arnold, J.N.; Roversi, P.; Crispin, M.D.; Radcliffe, C.; Lea, S.M.; Dwek, R.A.; Rudd, P.M.; Sim, R.B. Human complement factor I glycosylation: Structural and functional characterisation of the N-linked oligosaccharides. Biochim. Biophys. Acta 2006, 1764, 1757–1766. [Google Scholar] [CrossRef]
- Chan, W.H.; Hussain, A.A.; Marshall, J. Youngs Modulus of Bruchs Membrane: Implications for AMD. Investig. Ophthalmol. Vis. Sci. 2007, 48, 2187. [Google Scholar]
- Friberg, T.R.; Lace, J.W. A comparison of the elastic properties of human choroid and sclera. Exp. Eye Res. 1988, 47, 429–436. [Google Scholar] [CrossRef]
- Jones, I.L.; Warner, M.; Stevens, J.D. Mathematical modelling of the elastic properties of retina: A determination of Young’s modulus. Eye 1992, 6 Pt 6, 556–559. [Google Scholar] [CrossRef]
- Guo, L.; Hussain, A.A.; Limb, G.A.; Marshall, J. Age-dependent variation in metalloproteinase activity of isolated human Bruch’s membrane and choroid. Invest. Ophthalmol. Vis. Sci. 1999, 40, 2676–2682. [Google Scholar]
- Vranka, J.A.; Johnson, E.; Zhu, X.; Shepardson, A.; Alexander, J.P.; Bradley, J.M.; Wirtz, M.K.; Weleber, R.G.; Klein, M.L.; Acott, T.S. Discrete expression and distribution pattern of TIMP-3 in the human retina and choroid. Curr. Eye Res. 1997, 16, 102–110. [Google Scholar] [CrossRef]
- Sarks, S.H. Ageing and degeneration in the macular region: A clinico-pathological study. Br. J. Ophthalmol. 1976, 60, 324–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okubo, A.; Rosa, R.H., Jr.; Bunce, C.V.; Alexander, R.A.; Fan, J.T.; Bird, A.C.; Luthert, P.J. The relationships of age changes in retinal pigment epithelium and Bruch’s membrane. Investig. Ophthalmol. Vis. Sci. 1999, 40, 443–449. [Google Scholar]
- Ramrattan, R.S.; van der Schaft, T.L.; Mooy, C.M.; de Bruijn, W.C.; Mulder, P.G.; de Jong, P.T. Morphometric analysis of Bruch’s membrane, the choriocapillaris, and the choroid in aging. Investig. Ophthalmol. Vis. Sci. 1994, 35, 2857–2864. [Google Scholar]
- Pauleikhoff, D.; Wojteki, S.; Muller, D.; Bornfeld, N.; Heiligenhaus, A. Adhesive properties of basal membranes of Bruch’s membrane. Immunohistochemical studies of age-dependent changes in adhesive molecules and lipid deposits. Ophthalmologe 2000, 97, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Haimovici, R.; Gantz, D.L.; Rumelt, S.; Freddo, T.F.; Small, D.M. The lipid composition of drusen, Bruch’s membrane, and sclera by hot stage polarizing light microscopy. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1592–1599. [Google Scholar]
- Bird, A.C.; Marshall, J. Retinal pigment epithelial detachments in the elderly. Trans. Ophthalmol. Soc. UK 1986, 105 Pt 6, 674–682. [Google Scholar]
- Hussain, A.A.; Rowe, L.; Marshall, J. Age-related alterations in the diffusional transport of amino acids across the human Bruch’s-choroid complex. J. Opt. Soc. Am. A Opt. Image Sci. Vis. 2002, 19, 166–172. [Google Scholar] [CrossRef]
- Kamei, M.; Hollyfield, J.G. TIMP-3 in Bruch’s membrane: Changes during aging and in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 1999, 40, 2367–2375. [Google Scholar]
- Handa, J.T.; Verzijl, N.; Matsunaga, H.; Aotaki-Keen, A.; Lutty, G.A.; te Koppele, J.M.; Miyata, T.; Hjelmeland, L.M. Increase in the advanced glycation end product pentosidine in Bruch’s membrane with age. Investig. Ophthalmol. Vis. Sci. 1999, 40, 775–779. [Google Scholar]
- Pietkiewicz, J.; Seweryn, E.; Bartys, A.; Gamian, A. Receptors for advanced glycation end products and their physiological and clinical significance. Postepy Hig. Med. Dosw. 2008, 62, 511–523. [Google Scholar]
- Keenan, T.D.; Pickford, C.E.; Holley, R.J.; Clark, S.J.; Lin, W.; Dowsey, A.W.; Merry, C.L.; Day, A.J.; Bishop, P.N. Age-dependent changes in heparan sulfate in human Bruch’s membrane: Implications for age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5370–5379. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Higman, V.A.; Mulloy, B.; Perkins, S.J.; Lea, S.M.; Sim, R.B.; Day, A.J. His-384 allotypic variant of factor H associated with age-related macular degeneration has different heparin binding properties from the non-disease-associated form. J. Biol. Chem. 2006, 281, 24713–24720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prosser, B.E.; Johnson, S.; Roversi, P.; Herbert, A.P.; Blaum, B.S.; Tyrrell, J.; Jowitt, T.A.; Clark, S.J.; Tarelli, E.; Uhrin, D.; et al. Structural basis for complement factor H linked age-related macular degeneration. J. Exp. Med. 2007, 204, 2277–2283. [Google Scholar] [CrossRef] [Green Version]
- Davis, W.L.; Jones, R.G.; Hagler, H.K. An electron microscopic histochemical and analytical X-ray microprobe study of calcification in Bruch’s membrane from human eyes. J. Histochem. Cytochem. 1981, 29, 601–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ugarte, M.; Hussain, A.A.; Marshall, J. An experimental study of the elastic properties of the human Bruch’s membrane-choroid complex: Relevance to ageing. Br. J. Ophthalmol. 2006, 90, 621–626. [Google Scholar] [CrossRef] [Green Version]
- Moore, D.J.; Hussain, A.A.; Marshall, J. Age-related variation in the hydraulic conductivity of Bruch’s membrane. Investig. Ophthalmol. Vis. Sci. 1995, 36, 1290–1297. [Google Scholar]
- Starita, C.; Hussain, A.A.; Pagliarini, S.; Marshall, J. Hydrodynamics of ageing Bruch’s membrane: Implications for macular disease. Exp. Eye Res. 1996, 62, 565–572. [Google Scholar] [CrossRef]
- Starita, C.; Hussain, A.A.; Patmore, A.; Marshall, J. Localization of the site of major resistance to fluid transport in Bruch’s membrane. Investig. Ophthalmol. Vis. Sci. 1997, 38, 762–767. [Google Scholar]
- Hillenkamp, J.; Hussain, A.A.; Jackson, T.L.; Cunningham, J.R.; Marshall, J. The influence of path length and matrix components on ageing characteristics of transport between the choroid and the outer retina. Investig. Ophthalmol. Vis. Sci. 2004, 45, 1493–1498. [Google Scholar] [CrossRef] [Green Version]
- Moore, D.J.; Clover, G.M. The effect of age on the macromolecular permeability of human Bruch’s membrane. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2970–2975. [Google Scholar]
- Green, W.R.; Enger, C. Age-related macular degeneration histopathologic studies. The 1992 Lorenz E. Zimmerman Lecture. Ophthalmology 1993, 100, 1519–1535. [Google Scholar] [CrossRef]
- Curcio, C.A.; Presley, J.B.; Millican, C.L.; Medeiros, N.E. Basal deposits and drusen in eyes with age-related maculopathy: Evidence for solid lipid particles. Exp. Eye Res. 2005, 80, 761–775. [Google Scholar] [CrossRef] [PubMed]
- Crabb, J.W.; Miyagi, M.; Gu, X.; Shadrach, K.; West, K.A.; Sakaguchi, H.; Kamei, M.; Hasan, A.; Yan, L.; Rayborn, M.E.; et al. Drusen proteome analysis: An approach to the etiology of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 14682–14687. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, P.; Liew, G.; Gopinath, B.; Wong, T.Y. Age-related macular degeneration. Lancet 2018, 392, 1147–1159. [Google Scholar] [CrossRef]
- Sura, A.A.; Chen, L.; Messinger, J.D.; Swain, T.A.; McGwin, G., Jr.; Freund, K.B.; Curcio, C.A. Measuring the Contributions of Basal Laminar Deposit and Bruch’s Membrane in Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2020, 61, 19. [Google Scholar] [CrossRef]
- Sarks, S.; Cherepanoff, S.; Killingsworth, M.; Sarks, J. Relationship of Basal laminar deposit and membranous debris to the clinical presentation of early age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2007, 48, 968–977. [Google Scholar] [CrossRef]
- Penfold, P.L.; Madigan, M.C.; Gillies, M.C.; Provis, J.M. Immunological and aetiological aspects of macular degeneration. Prog. Retin. Eye Res. 2001, 20, 385–414. [Google Scholar] [CrossRef] [PubMed]
- Spaide, R.F.; Ooto, S.; Curcio, C.A. Subretinal drusenoid deposits AKA pseudodrusen. Surv. Ophthalmol. 2018, 63, 782–815. [Google Scholar] [CrossRef]
- Chakravarthy, U.; Wong, T.Y.; Fletcher, A.; Piault, E.; Evans, C.; Zlateva, G.; Buggage, R.; Pleil, A.; Mitchell, P. Clinical risk factors for age-related macular degeneration: A systematic review and meta-analysis. BMC Ophthalmol. 2010, 10, 31. [Google Scholar] [CrossRef] [Green Version]
- Shahid, H.; Khan, J.C.; Cipriani, V.; Sepp, T.; Matharu, B.K.; Bunce, C.; Harding, S.P.; Clayton, D.G.; Moore, A.T.; Yates, J.R.; et al. Age-related macular degeneration: The importance of family history as a risk factor. Br. J. Ophthalmol. 2012, 96, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Seddon, J.M.; Cote, J.; Page, W.F.; Aggen, S.H.; Neale, M.C. The US twin study of age-related macular degeneration: Relative roles of genetic and environmental influences. Arch. Ophthalmol. 2005, 123, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.J.; Zeiss, C.; Chew, E.Y.; Tsai, J.Y.; Sackler, R.S.; Haynes, C.; Henning, A.K.; SanGiovanni, J.P.; Mane, S.M.; Mayne, S.T.; et al. Complement factor H polymorphism in age-related macular degeneration. Science 2005, 308, 385–389. [Google Scholar] [CrossRef]
- Iyengar, S.K.; Song, D.; Klein, B.E.; Klein, R.; Schick, J.H.; Humphrey, J.; Millard, C.; Liptak, R.; Russo, K.; Jun, G.; et al. Dissection of genomewide-scan data in extended families reveals a major locus and oligogenic susceptibility for age-related macular degeneration. Am. J. Hum. Genet. 2004, 74, 20–39. [Google Scholar] [CrossRef] [Green Version]
- Fritsche, L.G.; Igl, W.; Bailey, J.N.; Grassmann, F.; Sengupta, S.; Bragg-Gresham, J.L.; Burdon, K.P.; Hebbring, S.J.; Wen, C.; Gorski, M.; et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 2016, 48, 134–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, T.W.; Grassmann, F.; Brandl, C.; Kiel, C.; Gunther, F.; Strunz, T.; Weidner, L.; Zimmermann, M.E.; Korb, C.A.; Poplawski, A.; et al. Genome-wide association meta-analysis for early age-related macular degeneration highlights novel loci and insights for advanced disease. BMC Med. Genom. 2020, 13, 120. [Google Scholar] [CrossRef]
- de Breuk, A.; Acar, I.E.; Kersten, E.; Schijvenaars, M.; Colijn, J.M.; Haer-Wigman, L.; Bakker, B.; de Jong, S.; Meester-Smoor, M.A.; Verzijden, T.; et al. Development of a Genotype Assay for Age-Related Macular Degeneration: The EYE-RISK Consortium. Ophthalmology 2021, 128, 1604–1617. [Google Scholar] [CrossRef]
- Tzoumas, N.; Kavanagh, D.; Cordell, H.J.; Lotery, A.J.; Patel, P.J.; Steel, D.H. Rare complement factor I variants associated with reduced macular thickness and age-related macular degeneration in the UK Biobank. Hum. Mol. Genet. 2022, 31, 2678–2692. [Google Scholar] [CrossRef]
- Khan, A.H.; Sutton, J.; Cree, A.J.; Khandhadia, S.; De Salvo, G.; Tobin, J.; Prakash, P.; Arora, R.; Amoaku, W.; Charbel Issa, P.; et al. Prevalence and phenotype associations of complement factor I mutations in geographic atrophy. Hum. Mutat. 2021, 42, 1139–1152. [Google Scholar] [CrossRef] [PubMed]
- Roth, F.; Bindewald, A.; Holz, F.G. Keypathophysiologic pathways in age-related macular disease. Graefes Arch. Clin. Exp. Ophthalmol. 2004, 242, 710–716. [Google Scholar] [CrossRef] [PubMed]
- Age-Related Eye Disease Study Research, G. Risk factors associated with age-related macular degeneration. A case-control study in the age-related eye disease study: Age-Related Eye Disease Study Report Number 3. Ophthalmology 2000, 107, 2224–2232. [Google Scholar] [CrossRef]
- Vingerling, J.R.; Hofman, A.; Grobbee, D.E.; de Jong, P.T. Age-related macular degeneration and smoking. The Rotterdam Study. Arch. Ophthalmol. 1996, 114, 1193–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delcourt, C.; Diaz, J.L.; Ponton-Sanchez, A.; Papoz, L. Smoking and age-related macular degeneration. The POLA Study. Pathologies Oculaires Liees a l’Age. Arch. Ophthalmol. 1998, 116, 1031–1035. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.R.; Lawrenson, J.G. Antioxidant vitamin and mineral supplements for slowing the progression of age-related macular degeneration. Cochrane Database Syst. Rev. 2017, 7, CD000254. [Google Scholar] [CrossRef]
- Gholami, P.; Lakshminarayanan, V. Optical Coherence Tomography Image Retinal Database. Available online: https://www.openicpsr.org/openicpsr/project/108503/version/V1/view (accessed on 2 April 2023).
- Staurenghi, G.; Sadda, S.; Chakravarthy, U.; Spaide, R.F.; International Nomenclature for Optical Coherence Tomography (IN•OCT) Panel. Proposed lexicon for anatomic landmarks in normal posterior segment spectral-domain optical coherence tomography: The IN*OCT consensus. Ophthalmology 2014, 121, 1572–1578. [Google Scholar] [CrossRef] [PubMed]
- Schottenhamml, J.; Moult, E.M.; Ploner, S.B.; Chen, S.; Novais, E.; Husvogt, L.; Duker, J.S.; Waheed, N.K.; Fujimoto, J.G.; Maier, A.K. OCT-OCTA segmentation: Combining structural and blood flow information to segment Bruch’s membrane. Biomed. Opt. Express 2021, 12, 84–99. [Google Scholar] [CrossRef]
- Cozzi, M.; Viola, F.; Belotti, M.; Cigada, M.; Cherepanoff, S.; Staurenghi, G.; Invernizzi, A. The In Vivo Correlation between Retinal Pigment Epithelium Thickness and Quantitative Fundus Autofluorescence in a White Population. Ophthalmol. Retin. 2021, 5, 365–373. [Google Scholar] [CrossRef]
- Chen, L.; Yang, P.; Curcio, C.A. Visualizing lipid behind the retina in aging and age-related macular degeneration, via indocyanine green angiography (ASHS-LIA). Eye 2022, 36, 1735–1746. [Google Scholar] [CrossRef]
- Lamin, A.; Oakley, J.D.; Dubis, A.M.; Russakoff, D.B.; Sivaprasad, S. Changes in volume of various retinal layers over time in early and intermediate age-related macular degeneration. Eye 2019, 33, 428–434. [Google Scholar] [CrossRef] [Green Version]
- Trinh, M.; Khou, V.; Kalloniatis, M.; Nivison-Smith, L. Location-Specific Thickness Patterns in Intermediate Age-Related Macular Degeneration Reveals Anatomical Differences in Multiple Retinal Layers. Investig. Ophthalmol. Vis. Sci. 2021, 62, 13. [Google Scholar] [CrossRef]
- Trinh, M.; Kalloniatis, M.; Alonso-Caneiro, D.; Nivison-Smith, L. High-Density Optical Coherence Tomography Analysis Provides Insights Into Early/Intermediate Age-Related Macular Degeneration Retinal Layer Changes. Investig. Ophthalmol. Vis. Sci. 2022, 63, 36. [Google Scholar] [CrossRef]
- Wykoff, C.C.; Rosenfeld, P.J.; Waheed, N.K.; Singh, R.P.; Ronca, N.; Slakter, J.S.; Staurenghi, G.; Mones, J.; Baumal, C.R.; Saroj, N.; et al. Characterizing New-Onset Exudation in the Randomized Phase 2 FILLY Trial of Complement Inhibitor Pegcetacoplan for Geographic Atrophy. Ophthalmology 2021, 128, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.N.; Borooah, S.; Lando, L.; Dans, K.; Mahroo, O.A.; Meshi, A.; Kalitzeos, A.; Agorogiannis, G.; Moghimi, S.; Freeman, W.R.; et al. Quantifying the Separation Between the Retinal Pigment Epithelium and Bruch’s Membrane using Optical Coherence Tomography in Patients with Inherited Macular Degeneration. Transl. Vis. Sci. Technol. 2020, 9, 26. [Google Scholar] [CrossRef] [PubMed]
- Walport, M.J. Complement first of two parts. N. Engl. J. Med. 2001, 344, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Morgan, B.P.; Walters, D.; Serna, M.; Bubeck, D. Terminal complexes of the complement system: New structural insights and their relevance to function. Immunol. Rev. 2016, 274, 141–151. [Google Scholar] [CrossRef]
- Morgan, B.P. The membrane attack complex as an inflammatory trigger. Immunobiology 2016, 221, 747–751. [Google Scholar] [CrossRef]
- Fields, M.A.; Del Priore, L.V.; Adelman, R.A.; Rizzolo, L.J. Interactions of the choroid, Bruch’s membrane, retinal pigment epithelium, and neurosensory retina collaborate to form the outer blood-retinal-barrier. Prog. Retin. Eye Res. 2020, 76, 100803. [Google Scholar] [CrossRef]
- Lejoyeux, R.; Benillouche, J.; Ong, J.; Errera, M.-H.; Rossi, E.A.; Singh, S.R.; Dansingani, K.K.; da Silva, S.; Sinha, D.; Sahel, J.-A.; et al. Choriocapillaris: Fundamentals and advancements. Prog. Retin. Eye Res. 2022, 87, 100997. [Google Scholar] [CrossRef]
- Mullins, R.F.; Johnson, M.N.; Faidley, E.A.; Skeie, J.M.; Huang, J. Choriocapillaris vascular dropout related to density of drusen in human eyes with early age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1606–1612. [Google Scholar] [CrossRef] [Green Version]
- Gerl, V.B.; Bohl, J.r.; Pitz, S.; Stoffelns, B.; Pfeiffer, N.; Bhakdi, S. Extensive deposits of complement C3d and C5b-9 in the choriocapillaris of eyes of patients with diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1104–1108. [Google Scholar]
- Hageman, G.S.; Anderson, D.H.; Johnson, L.V.; Hancox, L.S.; Taiber, A.J.; Hardisty, L.I.; Hageman, J.L.; Stockman, H.A.; Borchardt, J.D.; Gehrs, K.M. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 7227–7232. [Google Scholar] [CrossRef] [Green Version]
- Seth, A.; Cui, J.; To, E.; Kwee, M.; Matsubara, J. Complement-associated deposits in the human retina. Investig. Ophthalmol. Vis. Sci. 2008, 49, 743–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullins, R.F.; Dewald, A.D.; Streb, L.M.; Wang, K.; Kuehn, M.H.; Stone, E.M. Elevated membrane attack complex in human choroid with high risk complement factor H genotypes. Exp. Eye Res. 2011, 93, 565–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullins, R.F.; Schoo, D.P.; Sohn, E.H.; Flamme-Wiese, M.J.; Workamelahu, G.; Johnston, R.M.; Wang, K.; Tucker, B.A.; Stone, E.M. The membrane attack complex in aging human choriocapillaris: Relationship to macular degeneration and choroidal thinning. Am. J. Pathol. 2014, 184, 3142–3153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skeie, J.M.; Fingert, J.H.; Russell, S.R.; Stone, E.M.; Mullins, R.F. Complement component C5a activates ICAM-1 expression on human choroidal endothelial cells. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5336–5342. [Google Scholar] [CrossRef]
- Chirco, K.R.; Tucker, B.A.; Stone, E.M.; Mullins, R.F. Selective accumulation of the complement membrane attack complex in aging choriocapillaris. Exp. Eye Res. 2016, 146, 393–397. [Google Scholar] [CrossRef] [Green Version]
- Mulfaul, K.; Mullin, N.K.; Giacalone, J.C.; Voigt, A.P.; R DeVore, M.; Stone, E.M.; Tucker, B.A.; Mullins, R.F. Local factor H production by human choroidal endothelial cells mitigates complement deposition: Implications for macular degeneration. J. Pathol. 2022, 257, 29–38. [Google Scholar] [CrossRef]
- Whitmore, S.S.; Sohn, E.H.; Chirco, K.R.; Drack, A.V.; Stone, E.M.; Tucker, B.A.; Mullins, R.F. Complement activation and choriocapillaris loss in early AMD: Implications for pathophysiology and therapy. Prog. Retin. Eye Res. 2015, 45, 1–29. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimi, K.B.; Fijalkowski, N.; Cano, M.; Handa, J.T. Decreased membrane complement regulators in the retinal pigmented epithelium contributes to age-related macular degeneration. J. Pathol. 2013, 229, 729–742. [Google Scholar] [CrossRef] [Green Version]
- Anderson, D.H.; Radeke, M.J.; Gallo, N.B.; Chapin, E.A.; Johnson, P.T.; Curletti, C.R.; Hancox, L.S.; Hu, J.; Ebright, J.N.; Malek, G.; et al. The pivotal role of the complement system in aging and age-related macular degeneration: Hypothesis re-visited. Prog. Retin. Eye Res. 2010, 29, 95–112. [Google Scholar] [CrossRef] [Green Version]
- Menon, M.; Mohammadi, S.; Davila-Velderrain, J.; Goods, B.A.; Cadwell, T.D.; Xing, Y.; Stemmer-Rachamimov, A.; Shalek, A.K.; Love, J.C.; Kellis, M.; et al. Single-cell transcriptomic atlas of the human retina identifies cell types associated with age-related macular degeneration. Nat. Commun. 2019, 10, 4902. [Google Scholar] [CrossRef] [Green Version]
- Hageman, G.S.; Luthert, P.J.; Victor Chong, N.H.; Johnson, L.V.; Anderson, D.H.; Mullins, R.F. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog. Retin. Eye Res. 2001, 20, 705–732. [Google Scholar] [CrossRef]
- Baudouin, C.; Peyman, G.A.; Fredj-Reygrobellet, D.; Gordon, W.C.; Lapalus, P.; Gastaud, P.; Bazan, N.G. Immunohistological study of subretinal membranes in age-related macular degeneration. Jpn. J. Ophthalmol. 1992, 36, 443–451. [Google Scholar] [PubMed]
- Johnson, L.V.; Leitner, W.P.; Rivest, A.J.; Staples, M.K.; Radeke, M.J.; Anderson, D.H. The Alzheimer’s A beta -peptide is deposited at sites of complement activation in pathologic deposits associated with aging and age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 11830–11835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Godino, R.; Garland, D.L.; Pierce, E.A. A local complement response by RPE causes early-stage macular degeneration. Hum. Mol. Gen. 2015, 24, 5555–5569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garland, D.L.; Pierce, E.A.; Fernandez-Godino, R. Complement C5 is not critical for the formation of sub-RPE deposits in Efemp1 mutant mice. Sci. Rep. 2021, 11, 10416. [Google Scholar] [CrossRef]
- Crowley, M.A.; Garland, D.L.; Sellner, H.; Banks, A.; Fan, L.; Rejtar, T.; Buchanan, N.; Delgado, O.; Xu, Y.Y.; Jose, S.; et al. Complement factor B is critical for sub-RPE deposit accumulation in a model of Doyne honeycomb retinal dystrophy with features of age-related macular degeneration. Hum. Mol. Gen. 2023, 32, 204–217. [Google Scholar] [CrossRef]
- Ramkumar, H.L.; Zhang, J.; Chan, C.C. Retinal ultrastructure of murine models of dry age-related macular degeneration (AMD). Prog. Retin. Eye Res. 2010, 29, 169–190. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Muckersie, E.; Robertson, M.; Forrester, J.V.; Xu, H. Up-regulation of complement factor B in retinal pigment epithelial cells is accompanied by complement activation in the aged retina. Exp. Eye Res. 2008, 87, 543–550. [Google Scholar] [CrossRef]
- Chen, H.; Liu, B.; Lukas, T.J.; Neufeld, A.H. The aged retinal pigment epithelium/choroid: A potential substratum for the pathogenesis of age-related macular degeneration. PLoS ONE 2008, 3, e2339. [Google Scholar] [CrossRef]
- Wang, A.L.; Lukas, T.J.; Yuan, M.; Du, N.; Tso, M.O.; Neufeld, A.H. Autophagy and exosomes in the aged retinal pigment epithelium: Possible relevance to drusen formation and age-related macular degeneration. PLoS ONE 2009, 4, e4160. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.L.; Lukas, T.J.; Yuan, M.; Du, N.; Tso, M.O.; Neufeld, A.H. Autophagy, exosomes and drusen formation in age-related macular degeneration. Autophagy 2009, 5, 563–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Forrester, J.V.; Xu, H. Synthesis of complement factor H by retinal pigment epithelial cells is down-regulated by oxidized photoreceptor outer segments. Exp. Eye Res. 2007, 84, 635–645. [Google Scholar] [CrossRef]
- Kim, Y.H.; He, S.; Kase, S.; Kitamura, M.; Ryan, S.J.; Hinton, D.R. Regulated secretion of complement factor H by RPE and its role in RPE migration. Graefes Arch. Clin. Exp. Ophthalmol. 2009, 247, 651–659. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, B.J.; Fan, W.; Zheng, J.J.; Cai, H.; Del Priore, L.V.; Bora, N.S.; Kaplan, H.J. Novel role for a complement regulatory protein (CD46) in retinal pigment epithelial adhesion. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3669–3674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogt, S.D.; Barnum, S.R.; Curcio, C.A.; Read, R.W. Distribution of complement anaphylatoxin receptors and membrane-bound regulators in normal human retina. Exp. Eye Res. 2006, 83, 834–840. [Google Scholar] [CrossRef]
- Weismann, D.; Hartvigsen, K.; Lauer, N.; Bennett, K.L.; Scholl, H.P.N.; Issa, P.C.; Cano, M.; Brandstätter, H.; Tsimikas, S.; Skerka, C.; et al. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature 2011, 478, 76–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhutto, I.A.; Baba, T.; Merges, C.; Juriasinghani, V.; McLeod, D.S.; Lutty, G.A. C-reactive protein and complement factor H in aged human eyes and eyes with age-related macular degeneration. Br. J. Ophthalmol. 2011, 95, 1323–1330. [Google Scholar] [CrossRef] [Green Version]
- Vogt, S.D.; Curcio, C.A.; Wang, L.; Li, C.-M.; McGwin, G., Jr.; Medeiros, N.E.; Philp, N.J.; Kimble, J.A.; Read, R.W. Retinal pigment epithelial expression of complement regulator CD46 is altered early in the course of geographic atrophy. Exp. Eye Res. 2011, 93, 413–423. [Google Scholar] [CrossRef] [Green Version]
- Cerniauskas, E.; Kurzawa-Akanbi, M.; Xie, L.; Hallam, D.; Moya-Molina, M.; White, K.; Steel, D.; Doherty, M.; Whitfield, P.; Al-Aama, J.; et al. Complement modulation reverses pathology in Y402H-retinal pigment epithelium cell model of age-related macular degeneration by restoring lysosomal function. Stem Cells Transl. Med. 2020, 9, 1585–1603. [Google Scholar] [CrossRef]
- Ebeling, M.C.; Geng, Z.; Kapphahn, R.J.; Roehrich, H.; Montezuma, S.R.; Dutton, J.R.; Ferrington, D.A. Impaired Mitochondrial Function in iPSC-Retinal Pigment Epithelium with the Complement Factor H Polymorphism for Age-Related Macular Degeneration. Cells 2021, 10, 789. [Google Scholar] [CrossRef]
- Hallam, D.; Collin, J.; Bojic, S.; Chichagova, V.; Buskin, A.; Xu, Y.; Lafage, L.; Otten, E.G.; Anyfantis, G.; Mellough, C.; et al. An Induced Pluripotent Stem Cell Patient Specific Model of Complement Factor H (Y402H) Polymorphism Displays Characteristic Features of Age-Related Macular Degeneration and Indicates a Beneficial Role for UV Light Exposure. Stem Cells 2017, 35, 2305–2320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrington, D.A.; Kapphahn, R.J.; Leary, M.M.; Atilano, S.R.; Terluk, M.R.; Karunadharma, P.; Chen, G.K.-J.; Ratnapriya, R.; Swaroop, A.; Montezuma, S.R.; et al. Increased retinal mtDNA damage in the CFH variant associated with age-related macular degeneration. Exp. Eye Res. 2016, 145, 269–277. [Google Scholar] [CrossRef] [Green Version]
- Tan, L.X.; Toops, K.A.; Lakkaraju, A. Protective responses to sublytic complement in the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA 2016, 113, 8789–8794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armento, A.; Honisch, S.; Panagiotakopoulou, V.; Sonntag, I.; Jacob, A.; Bolz, S.; Kilger, E.; Deleidi, M.; Clark, S.; Ueffing, M. Loss of Complement Factor H impairs antioxidant capacity and energy metabolism of human RPE cells. Sci. Rep. 2020, 10, 10320. [Google Scholar] [CrossRef]
- Thurman, J.M.; Renner, B.; Kunchithapautham, K.; Ferreira, V.P.; Pangburn, M.K.; Ablonczy, Z.; Tomlinson, S.; Holers, V.M.; Rohrer, B. Oxidative stress renders retinal pigment epithelial cells susceptible to complement-mediated injury. J. Biol. Chem. 2009, 284, 16939–16947. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Lauer, T.W.; Sick, A.; Hackett, S.F.; Campochiaro, P.A. Oxidative stress modulates complement factor H expression in retinal pigmented epithelial cells by acetylation of FOXO3. J. Biol. Chem. 2007, 282, 22414–22425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, P.; Baciu, P.; Kerrigan, B.C.P.; Etheridge, M.; Sung, E.; Toimil, B.A.; Berchuck, J.E.; Jaffe, G.J. Retinal Pigment Epithelial Cell Death by the Alternative Complement Cascade: Role of Membrane Regulatory Proteins, Calcium, PKC, and Oxidative Stress. Investig. Ophthalmol. Vis. Sci. 2014, 55, 3012–3021. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimi, K.B.; Fijalkowski, N.; Cano, M.; Handa, J.T. Oxidized low-density-lipoprotein-induced injury in retinal pigment epithelium alters expression of the membrane complement regulatory factors CD46 and CD59 through exosomal and apoptotic bleb release. Adv. Exp. Med. Biol. 2014, 801, 259–265. [Google Scholar] [PubMed]
- Luo, C.; Zhao, J.; Madden, A.; Chen, M.; Xu, H. Complement expression in retinal pigment epithelial cells is modulated by activated macrophages. Exp. Eye Res. 2013, 112, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Toomey, C.B.; Kelly, U.; Saban, D.R.; Bowes Rickman, C. Regulation of age-related macular degeneration-like pathology by complement factor H. Proc. Natl. Acad. Sci. USA 2015, 112, E3040–E3049. [Google Scholar] [CrossRef] [Green Version]
- Lueck, K.; Wasmuth, S.; Williams, J.; Hughes, T.R.; Morgan, B.P.; Lommatzsch, A.; Greenwood, J.; Moss, S.E.; Pauleikhoff, D. Sub-lytic C5b-9 induces functional changes in retinal pigment epithelial cells consistent with age-related macular degeneration. Eye 2011, 25, 1074–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandyopadhyay, M.; Rohrer, B. Matrix metalloproteinase activity creates pro-angiogenic environment in primary human retinal pigment epithelial cells exposed to complement. Investig. Ophthalmol. Vis. Sci. 2012, 53, 1953–1961. [Google Scholar] [CrossRef] [Green Version]
- Sweigard, J.H.; Cashman, S.M.; Kumar-Singh, R. Adenovirus-mediated delivery of CD46 attenuates the alternative complement pathway on RPE: Implications for age-related macular degeneration. Gene Ther. 2011, 18, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Leaderer, D.; Cashman, S.M.; Kumar-Singh, R. Adeno-associated virus mediated delivery of an engineered protein that combines the complement inhibitory properties of CD46, CD55 and CD59. J. Gene Med. 2015, 17, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Cashman, S.M.; Ramo, K.; Kumar-Singh, R. A non membrane-targeted human soluble CD59 attenuates choroidal neovascularization in a model of age related macular degeneration. PLoS ONE 2011, 6, e19078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, A.; Mandwie, M.; Dreismann, A.K.; Smyth, C.M.; Doyle, H.; Malik, T.H.; Pickering, M.C.; Lachmann, P.J.; Alexander, I.E.; Logan, G.J. Adeno-Associated Virus Vector Gene Delivery Elevates Factor I Levels and Downregulates the Complement Alternative Pathway In Vivo. Hum. Gene Ther. 2021, 32, 1370–1381. [Google Scholar] [CrossRef]
- Schnabolk, G.; Coughlin, B.; Joseph, K.; Kunchithapautham, K.; Bandyopadhyay, M.; O’Quinn, E.C.; Nowling, T.; Rohrer, B. Local production of the alternative pathway component factor B is sufficient to promote laser-induced choroidal neovascularization. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1850–1863. [Google Scholar] [CrossRef] [Green Version]
- Rohrer, B.; Coughlin, B.; Kunchithapautham, K.; Long, Q.; Tomlinson, S.; Takahashi, K.; Holers, V.M. The alternative pathway is required, but not alone sufficient, for retinal pathology in mouse laser-induced choroidal neovascularization. Mol. Immunol. 2011, 48, e1–e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bora, N.S.; Kaliappan, S.; Jha, P.; Xu, Q.; Sohn, J.H.; Dhaulakhandi, D.B.; Kaplan, H.J.; Bora, P.S. Complement activation via alternative pathway is critical in the development of laser-induced choroidal neovascularization: Role of factor B and factor H. J. Immunol. 2006, 177, 1872–1878. [Google Scholar] [CrossRef] [Green Version]
- Bora, P.S.; Sohn, J.H.; Cruz, J.M.; Jha, P.; Nishihori, H.; Wang, Y.; Kaliappan, S.; Kaplan, H.J.; Bora, N.S. Role of complement and complement membrane attack complex in laser-induced choroidal neovascularization. J. Immunol. 2005, 174, 491–497. [Google Scholar] [CrossRef] [Green Version]
- Nozaki, M.; Raisler, B.J.; Sakurai, E.; Sarma, J.V.; Barnum, S.R.; Lambris, J.D.; Chen, Y.; Zhang, K.; Ambati, B.K.; Baffi, J.Z.; et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc. Natl. Acad. Sci. USA 2006, 103, 2328–2333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramo, K.; Cashman, S.M.; Kumar-Singh, R. Evaluation of adenovirus-delivered human CD59 as a potential therapy for AMD in a model of human membrane attack complex formation on murine RPE. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4126–4136. [Google Scholar] [CrossRef]
- Gandhi, J.; Cashman, S.M.; Kumar-Singh, R. Soluble CD59 expressed from an adenovirus in vivo is a potent inhibitor of complement deposition on murine liver vascular endothelium. PLoS ONE 2011, 6, e21621. [Google Scholar] [CrossRef] [PubMed]
- Bora, N.S.; Jha, P.; Lyzogubov, V.V.; Kaliappan, S.; Liu, J.; Tytarenko, R.G.; Fraser, D.A.; Morgan, B.P.; Bora, P.S. Recombinant membrane-targeted form of CD59 inhibits the growth of choroidal neovascular complex in mice. J. Biol. Chem. 2010, 285, 33826–33833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohrer, B.; Long, Q.; Coughlin, B.; Wilson, R.B.; Huang, Y.; Qiao, F.; Tang, P.H.; Kunchithapautham, K.; Gilkeson, G.S.; Tomlinson, S. A targeted inhibitor of the alternative complement pathway reduces angiogenesis in a mouse model of age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3056–3064. [Google Scholar] [CrossRef]
- Birke, K.; Lipo, E.; Birke, M.T.; Kumar-Singh, R. Topical application of PPADS inhibits complement activation and choroidal neovascularization in a model of age-related macular degeneration. PLoS ONE 2013, 8, e76766. [Google Scholar] [CrossRef] [Green Version]
- Bora, N.S.; Kaliappan, S.; Jha, P.; Xu, Q.; Sivasankar, B.; Harris, C.L.; Morgan, B.P.; Bora, P.S. CD59, a complement regulatory protein, controls choroidal neovascularization in a mouse model of wet-type age-related macular degeneration. J. Immunol. 2007, 178, 1783–1790. [Google Scholar] [CrossRef] [Green Version]
- Sweigard, J.H.; Yanai, R.; Gaissert, P.; Saint-Geniez, M.; Kataoka, K.; Thanos, A.; Stahl, G.L.; Lambris, J.D.; Connor, K.M. The alternative complement pathway regulates pathological angiogenesis in the retina. FASEB J. 2014, 28, 3171–3182. [Google Scholar] [CrossRef] [Green Version]
- Harboe, M.; Ulvund, G.; Vien, L.; Fung, M.; Mollnes, T.E. The quantitative role of alternative pathway amplification in classical pathway induced terminal complement activation. Clin. Exp. Immunol. 2004, 138, 439–446. [Google Scholar] [CrossRef]
- Kunchithapautham, K.; Rohrer, B. Sublytic membrane-attack-complex (MAC) activation alters regulated rather than constitutive vascular endothelial growth factor (VEGF) secretion in retinal pigment epithelium monolayers. J. Biol. Chem. 2011, 286, 23717–23724. [Google Scholar] [CrossRef] [Green Version]
- Lyzogubov, V.; Wu, X.; Jha, P.; Tytarenko, R.; Triebwasser, M.; Kolar, G.; Bertram, P.; Bora, P.S.; Atkinson, J.P.; Bora, N.S. Complement Regulatory Protein CD46 Protects against Choroidal Neovascularization in Mice. Am. J. Pathol. 2014, 184, 2537–2548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyzogubov, V.V.; Bora, P.S.; Wu, X.; Horn, L.E.; de Roque, R.; Rudolf, X.V.; Atkinson, J.P.; Bora, N.S. The Complement Regulatory Protein CD46 Deficient Mouse Spontaneously Develops Dry-Type Age-Related Macular Degeneration–Like Phenotype. Am. J. Pathol. 2016, 186, 2088–2104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keir, L.S.; Firth, R.; Aponik, L.; Feitelberg, D.; Sakimoto, S.; Aguilar, E.; Welsh, G.I.; Richards, A.; Usui, Y.; Satchell, S.C. VEGF regulates local inhibitory complement proteins in the eye and kidney. J. Clin. Investig. 2017, 127, 199–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahr, W.H. Complement halts angiogenesis gone wild. Blood 2010, 116, 4393–4394. [Google Scholar] [CrossRef] [Green Version]
- Langer, H.F.; Chung, K.J.; Orlova, V.V.; Choi, E.Y.; Kaul, S.; Kruhlak, M.J.; Alatsatianos, M.; DeAngelis, R.A.; Roche, P.A.; Magotti, P.; et al. Complement-mediated inhibition of neovascularization reveals a point of convergence between innate immunity and angiogenesis. Blood 2010, 116, 4395–4403. [Google Scholar] [CrossRef] [Green Version]
- Murray, H.; Qiu, B.; Ho, S.Y.; Wang, X. Complement Factor B Mediates Ocular Angiogenesis through Regulating the VEGF Signaling Pathway. Int. J. Mol. Sci. 2021, 22, 9580. [Google Scholar] [CrossRef]
- Zeng, S.; Whitmore, S.S.; Sohn, E.H.; Riker, M.J.; Wiley, L.A.; Scheetz, T.E.; Stone, E.M.; Tucker, B.A.; Mullins, R.F. Molecular response of chorioretinal endothelial cells to complement injury: Implications for macular degeneration. J. Pathol. 2016, 238, 446–456. [Google Scholar] [CrossRef] [Green Version]
- Tzoumas, N.R.G.; Williams, M.A.; Steel, D.H. Complement inhibitors for age-related macular degeneration. Cochrane Database Syst. Rev. 2023; in press. [Google Scholar] [CrossRef]
- Hanna, R.M.; Barsoum, M.; Arman, F.; Selamet, U.; Hasnain, H.; Kurtz, I. Nephrotoxicity induced by intravitreal vascular endothelial growth factor inhibitors: Emerging evidence. Kidney Int. 2019, 96, 572–580. [Google Scholar] [CrossRef]
- Saint-Geniez, M.; Kurihara, T.; Sekiyama, E.; Maldonado, A.E.; D’Amore, P.A. An essential role for RPE-derived soluble VEGF in the maintenance of the choriocapillaris. Proc. Natl. Acad. Sci. USA 2009, 106, 18751–18756. [Google Scholar] [CrossRef] [Green Version]
- Grunwald, J.E.; Daniel, E.; Huang, J.; Ying, G.-s.; Maguire, M.G.; Toth, C.A.; Jaffe, G.J.; Fine, S.L.; Blodi, B.; Klein, M.L.; et al. Risk of Geographic Atrophy in the Comparison of Age-related Macular Degeneration Treatments Trials. Ophthalmology 2014, 121, 150–161. [Google Scholar] [CrossRef] [Green Version]
- Young, M.; Chui, L.; Fallah, N.; Or, C.; Merkur, A.B.; Kirker, A.W.; Albiani, D.A.; Forooghian, F. Exacerbation of choroidal and retinal pigment epithelial atrophy after anti-vascular endothelial growth factor treatment in neovascular age-related macular degeneration. Retina 2014, 34, 1308–1315. [Google Scholar] [CrossRef]
- Xu, L.; Mrejen, S.; Jung, J.J.; Gallego-Pinazo, R.; Thompson, D.; Marsiglia, M.; Freund, K.B. Geographic atrophy in patients receiving anti-vascular endothelial growth factor for neovascular age-related macular degeneration. Retina 2015, 35, 176–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenfeld, P.J.; Shapiro, H.; Tuomi, L.; Webster, M.; Elledge, J.; Blodi, B. Characteristics of Patients Losing Vision after 2 Years of Monthly Dosing in the Phase III Ranibizumab Clinical Trials. Ophthalmology 2011, 118, 523–530. [Google Scholar] [CrossRef]
- Pfau, M.; Möller, P.T.; Künzel, S.H.; von der Emde, L.; Lindner, M.; Thiele, S.; Dysli, C.; Nadal, J.; Schmid, M.; Schmitz-Valckenberg, S.; et al. Type 1 Choroidal Neovascularization Is Associated with Reduced Localized Progression of Atrophy in Age-Related Macular Degeneration. Ophthalmol. Retin. 2020, 4, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Capuano, V.; Miere, A.; Querques, L.; Sacconi, R.; Carnevali, A.; Amoroso, F.; Bandello, F.; Souied, E.H.; Querques, G. Treatment-naïve quiescent choroidal neovascularization in geographic atrophy secondary to nonexudative age-related macular degeneration. Am. J. Ophthalmol. 2017, 182, 45–55. [Google Scholar] [CrossRef]
- Heiferman, M.J.; Fawzi, A.A. Progression of subclinical choroidal neovascularization in age-related macular degeneration. PLoS ONE 2019, 14, e0217805. [Google Scholar] [CrossRef] [Green Version]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roumenina, L.T.; Daugan, M.V.; Petitprez, F.; Sautès-Fridman, C.; Fridman, W.H. Context-dependent roles of complement in cancer. Nat. Rev. Cancer 2019, 19, 698–715. [Google Scholar] [CrossRef] [Green Version]
- Wood, A.J.T.; Vassallo, A.; Summers, C.; Chilvers, E.R.; Conway-Morris, A. C5a anaphylatoxin and its role in critical illness-induced organ dysfunction. Eur. J. Clin. Investig. 2018, 48, e13028. [Google Scholar] [CrossRef] [Green Version]
- Triantafilou, M.; Hughes, T.R.; Morgan, B.P.; Triantafilou, K. Complementing the inflammasome. Immunology 2016, 147, 152–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strainic, M.G.; Liu, J.; Huang, D.; An, F.; Lalli, P.N.; Muqim, N.; Shapiro, V.S.; Dubyak, G.R.; Heeger, P.S.; Medof, M.E. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity 2008, 28, 425–435. [Google Scholar] [CrossRef] [Green Version]
- Peng, Q.; Li, K.; Wang, N.; Li, Q.; Asgari, E.; Lu, B.; Woodruff, T.M.; Sacks, S.H.; Zhou, W. Dendritic cell function in allostimulation is modulated by C5aR signaling. J. Immunol. 2009, 183, 6058–6068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piao, C.; Zhang, W.-M.; Li, T.-T.; Zhang, C.-c.; Qiu, S.; Liu, Y.; Liu, S.; Jin, M.; Jia, L.-X.; Song, W.-C. Complement 5a stimulates macrophage polarization and contributes to tumor metastases of colon cancer. Exp. Cell. Res. 2018, 366, 127–138. [Google Scholar] [CrossRef]
- Han, G.; Geng, S.; Li, Y.; Chen, G.; Wang, R.; Li, X.; Ma, Y.; Shen, B.; Li, Y. γδT-cell function in sepsis is modulated by C5a receptor signalling. Immunology 2011, 133, 340–349. [Google Scholar] [CrossRef]
- Zaal, A.; Lissenberg-Thunnissen, S.N.; van Schijndel, G.; Wouters, D.; van Ham, S.M.; ten Brinke, A. Crosstalk between Toll like receptors and C5a receptor in human monocyte derived DCs suppress inflammatory cytokine production. Immunobiology 2013, 218, 175–180. [Google Scholar] [CrossRef]
- Li, K.; Fazekasova, H.; Wang, N.; Peng, Q.; Sacks, S.H.; Lombardi, G.; Zhou, W. Functional modulation of human monocytes derived DCs by anaphylatoxins C3a and C5a. Immunobiology 2012, 217, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Li, X.X.; Clark, R.J.; Woodruff, T.M. C5aR2 Activation Broadly Modulates the Signaling and Function of Primary Human Macrophages. J. Immunol. 2020, 205, 1102–1112. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, B.; Schnabolk, G.; Joseph, K.; Raikwar, H.; Kunchithapautham, K.; Johnson, K.; Moore, K.; Wang, Y.; Rohrer, B. Connecting the innate and adaptive immune responses in mouse choroidal neovascularization via the anaphylatoxin C5a and γδT-cells. Sci. Rep. 2016, 6, 23794. [Google Scholar] [CrossRef] [Green Version]
- Ribot, J.C.; Lopes, N.; Silva-Santos, B. γδ T cells in tissue physiology and surveillance. Nat. Rev. Immunol. 2021, 21, 221–232. [Google Scholar] [CrossRef]
- Jane-Wit, D.; Manes, T.D.; Yi, T.; Qin, L.; Clark, P.; Kirkiles-Smith, N.C.; Abrahimi, P.; Devalliere, J.; Moeckel, G.; Kulkarni, S. Alloantibody and complement promote T cell–mediated cardiac allograft vasculopathy through noncanonical nuclear factor-κB signaling in endothelial cells. Circulation 2013, 128, 2504–2516. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.B.; Qin, L.; Li, G.; Fang, C.; Kirkiles-Smith, N.C.; Tellides, G.; Pober, J.S.; Jane-Wit, D. Complement membrane attack complexes assemble NLRP3 inflammasomes triggering IL-1 activation of IFN-γ–primed human endothelium. Circ. Res. 2019, 124, 1747–1759. [Google Scholar] [CrossRef]
- Kilgore, K.S.; Schmid, E.; Shanley, T.P.; Flory, C.M.; Maheswari, V.; Tramontini, N.L.; Cohen, H.; Ward, P.A.; Friedl, H.P.; Warren, J.S. Sublytic concentrations of the membrane attack complex of complement induce endothelial interleukin-8 and monocyte chemoattractant protein-1 through nuclear factor-kappa B activation. Am. J. Pathol. 1997, 150, 2019. [Google Scholar] [PubMed]
- Brunn, G.J.; Saadi, S.; Platt, J.L. Differential regulation of endothelial cell activation by complement and interleukin 1α. Circ. Res. 2006, 98, 793–800. [Google Scholar] [CrossRef] [Green Version]
- Saadi, S.; Holzknecht, R.A.; Patte, C.P.; Platt, J.L. Endothelial cell activation by pore-forming structures: Pivotal role for interleukin-1α. Circulation 2000, 101, 1867–1873. [Google Scholar] [CrossRef] [Green Version]
- Copland, D.A.; Theodoropoulou, S.; Liu, J.; Dick, A.D. A Perspective of AMD Through the Eyes of Immunology. Investig. Ophthalmol. Vis. Sci. 2018, 59, AMD83–AMD92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrera, A.P.; Bhaskaran, A.; Xu, J.; Yang, X.; Scott, H.A.; Mohideen, U.; Ghosh, K. Senescence Increases Choroidal Endothelial Stiffness and Susceptibility to Complement Injury: Implications for Choriocapillaris Loss in AMD. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5910–5918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calippe, B.; Augustin, S.; Beguier, F.; Charles-Messance, H.; Poupel, L.; Conart, J.-B.; Hu, S.J.; Lavalette, S.; Fauvet, A.; Rayes, J.; et al. Complement Factor H Inhibits CD47-Mediated Resolution of Inflammation. Immunity 2017, 46, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Sacks, S.H.; Zhou, W. The relative importance of local and systemic complement production in ischaemia, transplantation and other pathologies. Mol. Immunol. 2007, 44, 3866–3874. [Google Scholar] [CrossRef]
- Kouser, L.; Abdul-Aziz, M.; Nayak, A.; Stover, C.M.; Sim, R.B.; Kishore, U. Properdin and factor h: Opposing players on the alternative complement pathway “see-saw”. Front. Immunol. 2013, 4, 93. [Google Scholar] [CrossRef] [Green Version]
- Laufer, J.; Katz, Y.; Passwell, J.H. Extrahepatic synthesis of complement proteins in inflammation. Mol. Immunol. 2001, 38, 221–229. [Google Scholar] [CrossRef]
- Sheerin, N.S.; Abe, K.; Risley, P.; Sacks, S.H. Accumulation of immune complexes in glomerular disease is independent of locally synthesized c3. J. Am. Soc. Nephrol. 2006, 17, 686–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.W.; Wong, T.Y.; Cheng, C.-Y.; Sabanayagam, C. Kidney and eye diseases: Common risk factors, etiological mechanisms, and pathways. Kidney Int. 2014, 85, 1290–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duvall-Young, J.; MacDonald, M.K.; McKechnie, N.M. Fundus changes in (type II) mesangiocapillary glomerulonephritis simulating drusen: A histopathological report. Br. J. Ophthalmol. 1989, 73, 297–302. [Google Scholar] [CrossRef]
- Dopler, A.; Guntau, L.; Harder, M.J.; Palmer, A.; Höchsmann, B.; Schrezenmeier, H.; Simmet, T.; Huber-Lang, M.; Schmidt, C.Q. Self versus nonself discrimination by the soluble complement regulators factor H and FHL-1. J. Immunol. 2019, 202, 2082–2094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallam, T.M.; Marchbank, K.J.; Harris, C.L.; Osmond, C.; Shuttleworth, V.G.; Griffiths, H.; Cree, A.J.; Kavanagh, D.; Lotery, A.J. Rare genetic variants in complement factor I lead to low FI plasma levels resulting in increased risk of age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2020, 61, 18. [Google Scholar] [CrossRef]
- Reynolds, R.; Hartnett, M.E.; Atkinson, J.P.; Giclas, P.C.; Rosner, B.; Seddon, J.M. Plasma complement components and activation fragments: Associations with age-related macular degeneration genotypes and phenotypes. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5818–5827. [Google Scholar] [CrossRef] [Green Version]
- Scholl, H.P.; Issa, P.C.; Walier, M.; Janzer, S.; Pollok-Kopp, B.; Börncke, F.; Fritsche, L.G.; Chong, N.V.; Fimmers, R.; Wienker, T. Systemic complement activation in age-related macular degeneration. PLoS ONE 2008, 3, e2593. [Google Scholar] [CrossRef]
- Lin, J.B.; Serghiou, S.; Miller, J.W.; Vavvas, D.G. Systemic Complement Activation Profiles in Nonexudative Age-Related Macular Degeneration: A Systematic Review. Ophthalmol. Sci. 2022, 2, 100118. [Google Scholar] [CrossRef]
- Heesterbeek, T.J.; Lechanteur, Y.T.; Lorés-Motta, L.; Schick, T.; Daha, M.R.; Altay, L.; Liakopoulos, S.; Smailhodzic, D.; den Hollander, A.I.; Hoyng, C.B. Complement activation levels are related to disease stage in AMD. Investig. Ophthalmol. Vis. Sci. 2020, 61, 18. [Google Scholar] [CrossRef] [Green Version]
- Lorés-Motta, L.; Paun, C.C.; Corominas, J.; Pauper, M.; Geerlings, M.J.; Altay, L.; Schick, T.; Daha, M.R.; Fauser, S.; Hoyng, C.B. Genome-wide association study reveals variants in CFH and CFHR4 associated with systemic complement activation: Implications in age-related macular degeneration. Ophthalmology 2018, 125, 1064–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, A.M.; Mandava, N.; Patnaik, J.L.; Frazer-Abel, A.A.; Wagner, B.D.; Palestine, A.G.; Mathias, M.T.; Siringo, F.S.; Cathcart, J.N.; Holers, V.M. Systemic activation of the complement system in patients with advanced age-related macular degeneration. Eur. J. Ophthalmol. 2020, 30, 1061–1068. [Google Scholar] [CrossRef]
- Balmforth, C.; van Bragt, J.J.; Ruijs, T.; Cameron, J.R.; Kimmitt, R.; Moorhouse, R.; Czopek, A.; Hu, M.K.; Gallacher, P.J.; Dear, J.W. Chorioretinal thinning in chronic kidney disease links to inflammation and endothelial dysfunction. JCI insight 2016, 1, e89173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergen, A.A.; Arya, S.; Koster, C.; Pilgrim, M.G.; Wiatrek-Moumoulidis, D.; van der Spek, P.J.; Hauck, S.M.; Boon, C.J.; Emri, E.; Stewart, A.J. On the origin of proteins in human drusen: The meet, greet and stick hypothesis. Prog. Retin. Eye Res. 2019, 70, 55–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kavanagh, D.; Yu, Y.; Schramm, E.C.; Triebwasser, M.; Wagner, E.K.; Raychaudhuri, S.; Daly, M.J.; Atkinson, J.P.; Seddon, J.M. Rare genetic variants in the CFI gene are associated with advanced age-related macular degeneration and commonly result in reduced serum factor I levels. Hum. Mol. Genet. 2015, 24, 3861–3870. [Google Scholar] [CrossRef] [Green Version]
- Wong, E.K.S.; Kavanagh, D. Diseases of complement dysregulation-an overview. Semin. Immunopathol. 2018, 40, 49–64. [Google Scholar] [CrossRef] [Green Version]
- Raychaudhuri, S.; Iartchouk, O.; Chin, K.; Tan, P.L.; Tai, A.K.; Ripke, S.; Gowrisankar, S.; Vemuri, S.; Montgomery, K.; Yu, Y.; et al. A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nat. Genet. 2011, 43, 1232–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Recalde, S.; Tortajada, A.; Subias, M.; Anter, J.; Blasco, M.; Maranta, R.; Coco, R.; Pinto, S.; Noris, M.; Garcia-Layana, A.; et al. Molecular Basis of Factor H R1210C Association with Ocular and Renal Diseases. J. Am. Soc. Nephrol. 2016, 27, 1305–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khandhadia, S.; Hakobyan, S.; Heng, L.Z.; Gibson, J.; Adams, D.H.; Alexander, G.J.; Gibson, J.M.; Martin, K.R.; Menon, G.; Nash, K.; et al. Age-related Macular Degeneration and Modification of Systemic Complement Factor H Production Through Liver Transplantation. Ophthalmology 2013, 120, 1612–1618. [Google Scholar] [CrossRef]
- Harris, C.L. Expanding horizons in complement drug discovery: Challenges and emerging strategies. Semin. Immunopathol. 2018, 40, 125–140. [Google Scholar] [CrossRef] [Green Version]
- Mastellos, D.C.; Ricklin, D.; Lambris, J.D. Clinical promise of next-generation complement therapeutics. Nat. Rev. Drug. Discov. 2019, 18, 707–729. [Google Scholar] [CrossRef]
- Bill, A.; Sperber, G.O. Control of retinal and choroidal blood flow. Eye 1990, 4, 319–325. [Google Scholar] [CrossRef] [PubMed]
- del Amo, E.M.; Rimpelä, A.-K.; Heikkinen, E.; Kari, O.K.; Ramsay, E.; Lajunen, T.; Schmitt, M.; Pelkonen, L.; Bhattacharya, M.; Richardson, D.; et al. Pharmacokinetic aspects of retinal drug delivery. Prog. Retin. Eye Res. 2017, 57, 134–185. [Google Scholar] [CrossRef]
- Pitkänen, L.; Ranta, V.-P.; Moilanen, H.; Urtti, A. Permeability of Retinal Pigment Epithelium: Effects of Permeant Molecular Weight and Lipophilicity. Investig. Ophthalmol. Vis. Sci. 2005, 46, 641–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vellonen, K.-S.; Soini, E.-M.; del Amo, E.M.; Urtti, A. Prediction of Ocular Drug Distribution from Systemic Blood Circulation. Mol. Pharm. 2016, 13, 2906–2911. [Google Scholar] [CrossRef] [PubMed]
- Hillmen, P.; Szer, J.; Weitz, I.; Röth, A.; Höchsmann, B.; Panse, J.; Usuki, K.; Griffin, M.; Kiladjian, J.-J.; de Castro, C. Pegcetacoplan versus eculizumab in paroxysmal nocturnal hemoglobinuria. N. Engl. J. Med. 2021, 384, 1028–1037. [Google Scholar] [CrossRef]
- McKinley, C.E.; Richards, S.J.; Munir, T.; Griffin, M.; Mitchell, L.D.; Arnold, L.; Riley, K.; Copeland, N.; Newton, D.J.; Hill, A. Extravascular hemolysis due to C3-loading in patients with PNH treated with eculizumab: Defining the clinical syndrome. Blood 2017, 130, 3471. [Google Scholar]
- Jayne, D.R.W.; Merkel, P.A.; Schall, T.J.; Bekker, P.; Group, A.S. Avacopan for the Treatment of ANCA-Associated Vasculitis. N. Engl. J. Med. 2021, 384, 599–609. [Google Scholar] [CrossRef]
- Risitano, A.M.; Roth, A.; Soret, J.; Frieri, C.; de Fontbrune, F.S.; Marano, L.; Alashkar, F.; Benajiba, L.; Marotta, S.; Rozenberg, I.; et al. Addition of iptacopan, an oral factor B inhibitor, to eculizumab in patients with paroxysmal nocturnal haemoglobinuria and active haemolysis: An open-label, single-arm, phase 2, proof-of-concept trial. Lancet Haematol. 2021, 8, e344–e354. [Google Scholar] [CrossRef]
- Yehoshua, Z.; Alexandre de Amorim Garcia Filho, C.; Nunes, R.P.; Gregori, G.; Penha, F.M.; Moshfeghi, A.A.; Zhang, K.; Sadda, S.; Feuer, W.; Rosenfeld, P.J. Systemic Complement Inhibition with Eculizumab for Geographic Atrophy in Age-Related Macular Degeneration: The COMPLETE Study. Ophthalmology 2014, 121, 693–701. [Google Scholar] [CrossRef] [Green Version]
- Boyer, D.D.; Ko, Y.-P.; Podos, S.D.; Cartwright, M.E.; Gao, X.; Wiles, J.A.; Huang, M. Danicopan, an Oral Complement Factor D Inhibitor, Exhibits High and Sustained Exposure in Ocular Tissues in Preclinical Studies. Transl. Vis. Sci. Technol. 2022, 11, 37. [Google Scholar] [CrossRef] [PubMed]
- Sampat, K.M.; Garg, S.J. Complications of intravitreal injections. Curr. Opin. Ophthalmol. 2010, 21, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.S.; Grossi, F.V.; El Mehdi, D.; Gerber, M.R.; Brown, D.M.; Heier, J.S.; Wykoff, C.C.; Singerman, L.J.; Abraham, P.; Grassmann, F.; et al. Complement C3 Inhibitor Pegcetacoplan for Geographic Atrophy Secondary to Age-Related Macular Degeneration: A Randomized Phase 2 Trial. Ophthalmology 2020, 127, 186–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdera, H.C.; Kuranda, K.; Mingozzi, F. AAV Vector Immunogenicity in Humans: A Long Journey to Successful Gene Transfer. Mol. Ther. 2020, 28, 723–746. [Google Scholar] [CrossRef]
- Van Craenenbroeck, K.; Vanhoenacker, P.; Haegeman, G. Episomal vectors for gene expression in mammalian cells. Eur. J. Biochem. 2000, 267, 5665–5678. [Google Scholar] [CrossRef]
- Leroy, B.P.; Fischer, M.D.; Flannery, J.G.; MacLaren, R.E.; Dalkara, D.; Scholl, H.P.N.; Chung, D.C.; Spera, C.; Viriato, D.; Banhazi, J. Gene therapy for inherited retinal disease: Long-term durability of effect. Ophthalmic Res. 2022, 66, 179–196. [Google Scholar] [CrossRef]
- Trapani, I.; Colella, P.; Sommella, A.; Iodice, C.; Cesi, G.; de Simone, S.; Marrocco, E.; Rossi, S.; Giunti, M.; Palfi, A.; et al. Effective delivery of large genes to the retina by dual AAV vectors. EMBO Mol. Med. 2014, 6, 194–211. [Google Scholar] [CrossRef]
- Newman, N.J.; Yu-Wai-Man, P.; Carelli, V.; Moster, M.L.; Biousse, V.; Vignal-Clermont, C.; Sergott, R.C.; Klopstock, T.; Sadun, A.A.; Barboni, P.; et al. Efficacy and Safety of Intravitreal Gene Therapy for Leber Hereditary Optic Neuropathy Treated within 6 Months of Disease Onset. Ophthalmology 2021, 128, 649–660. [Google Scholar] [CrossRef]
- Dalkara, D.; Kolstad, K.D.; Caporale, N.; Visel, M.; Klimczak, R.R.; Schaffer, D.V.; Flannery, J.G. Inner limiting membrane barriers to AAV-mediated retinal transduction from the vitreous. Mol. Ther. 2009, 17, 2096–2102. [Google Scholar] [CrossRef] [Green Version]
- Hellstrom, M.; Ruitenberg, M.J.; Pollett, M.A.; Ehlert, E.M.; Twisk, J.; Verhaagen, J.; Harvey, A.R. Cellular tropism and transduction properties of seven adeno-associated viral vector serotypes in adult retina after intravitreal injection. Gene Ther. 2009, 16, 521–532. [Google Scholar] [CrossRef] [Green Version]
- Dalkara, D.; Byrne, L.C.; Klimczak, R.R.; Visel, M.; Yin, L.; Merigan, W.H.; Flannery, J.G.; Schaffer, D.V. In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci. Transl. Med. 2013, 5, 189ra176. [Google Scholar] [CrossRef]
- Ross, M.; Ofri, R. The future of retinal gene therapy: Evolving from subretinal to intravitreal vector delivery. Neural Regen. Res. 2021, 16, 1751–1759. [Google Scholar] [PubMed]
- Boye, S.L.; Bennett, A.; Scalabrino, M.L.; McCullough, K.T.; Van Vliet, K.; Choudhury, S.; Ruan, Q.; Peterson, J.; Agbandje-McKenna, M.; Boye, S.E. Impact of Heparan Sulfate Binding on Transduction of Retina by Recombinant Adeno-Associated Virus Vectors. J. Virol. 2016, 90, 4215–4231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodard, K.T.; Liang, K.J.; Bennett, W.C.; Samulski, R.J. Heparan Sulfate Binding Promotes Accumulation of Intravitreally Delivered Adeno-associated Viral Vectors at the Retina for Enhanced Transduction but Weakly Influences Tropism. J. Virol. 2016, 90, 9878–9888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.; Zeng, Y.; Sardar Pasha, S.P.B.; Bush, R.A.; Vijayasarathy, C.; Qian, H.; Wei, L.; Wiley, H.E.; Wu, Z.; Sieving, P.A. Trans-Ocular Electric Current In Vivo Enhances AAV-Mediated Retinal Transduction in Large Animal Eye After Intravitreal Vector Administration. Transl. Vis. Sci. Technol. 2020, 9, 28. [Google Scholar] [CrossRef]
- Ross, M.; Obolensky, A.; Averbukh, E.; Ezra-Elia, R.; Yamin, E.; Honig, H.; Dvir, H.; Rosov, A.; Hauswirth, W.W.; Gootwine, E. Evaluation of photoreceptor transduction efficacy of capsid-modified adeno-associated viral vectors following intravitreal and subretinal delivery in sheep. Hum. Gene Ther. 2020, 31, 719–729. [Google Scholar] [CrossRef]
- Teo, K.Y.C.; Lee, S.Y.; Barathi, A.V.; Tun, S.B.B.; Tan, L.; Constable, I.J. Surgical removal of internal limiting membrane and layering of AAV vector on the retina under air enhances gene transfection in a nonhuman primate. Investig. Ophthalmol. Vis. Sci. 2018, 59, 3574–3583. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Igarashi, T.; Miyake, K.; Kobayashi, M.; Yaguchi, C.; Iijima, O.; Yamazaki, Y.; Katakai, Y.; Miyake, N.; Kameya, S. Improved intravitreal AAV-mediated inner retinal gene transduction after surgical internal limiting membrane peeling in cynomolgus monkeys. Mol. Ther. 2017, 25, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Boye, S.E.; Alexander, J.J.; Witherspoon, C.D.; Boye, S.L.; Peterson, J.J.; Clark, M.E.; Sandefer, K.J.; Girkin, C.A.; Hauswirth, W.W.; Gamlin, P.D. Highly Efficient Delivery of Adeno-Associated Viral Vectors to the Primate Retina. Hum. Gene Ther. 2016, 27, 580–597. [Google Scholar] [CrossRef] [Green Version]
- Gamlin, P.D.; Alexander, J.J.; Boye, S.L.; Witherspoon, C.D.; Boye, S.E. SubILM injection of AAV for gene delivery to the retina. In Adeno-Associated Virus Vectors. Methods in Molecular Biology; Humana Press: New York, NY, USA, 2019; pp. 249–262. [Google Scholar]
- Peynshaert, K.; Vanluchene, H.; De Clerck, K.; Minnaert, A.-K.; Verhoeven, M.; Gouspillou, N.; Bostan, N.; Hisatomi, T.; Accou, G.; Sauvage, F.; et al. ICG-mediated photodisruption of the inner limiting membrane enhances retinal drug delivery. J. Control. Release 2022, 349, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.S.; Lee, V.; Wei, Z.; Song, J.Y.; Casal, G.; Cronin, T.; Willett, K.; Huckfeldt, R.; Morgan, J.I.; Aleman, T.S.; et al. Evaluation of Dose and Safety of AAV7m8 and AAV8BP2 in the Non-Human Primate Retina. Hum. Gene Ther. 2017, 28, 154–167. [Google Scholar] [CrossRef] [Green Version]
- Reichel, F.F.; Dauletbekov, D.L.; Klein, R.; Peters, T.; Ochakovski, G.A.; Seitz, I.P.; Wilhelm, B.; Ueffing, M.; Biel, M.; Wissinger, B.; et al. AAV8 Can Induce Innate and Adaptive Immune Response in the Primate Eye. Mol. Ther. 2017, 25, 2648–2660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichel, F.F.; Peters, T.; Wilhelm, B.; Biel, M.; Ueffing, M.; Wissinger, B.; Bartz-Schmidt, K.U.; Klein, R.; Michalakis, S.; Fischer, M.D.; et al. Humoral Immune Response After Intravitreal But Not After Subretinal AAV8 in Primates and Patients. Investig. Ophthalmol. Vis. Sci. 2018, 59, 1910–1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seitz, I.P.; Michalakis, S.; Wilhelm, B.; Reichel, F.F.; Ochakovski, G.A.; Zrenner, E.; Ueffing, M.; Biel, M.; Wissinger, B.; Bartz-Schmidt, K.U.; et al. Superior Retinal Gene Transfer and Biodistribution Profile of Subretinal Versus Intravitreal Delivery of AAV8 in Nonhuman Primates. Investig. Ophthalmol. Vis. Sci. 2017, 58, 5792–5801. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Miller, R.; Han, P.-Y.; Pang, J.; Dinculescu, A.; Chiodo, V.; Hauswirth, W.W. Intraocular route of AAV2 vector administration defines humoral immune response and therapeutic potential. Mol. Vis. 2008, 14, 1760–1769. [Google Scholar]
- Kotterman, M.A.; Yin, L.; Strazzeri, J.M.; Flannery, J.G.; Merigan, W.H.; Schaffer, D.V. Antibody neutralization poses a barrier to intravitreal adeno-associated viral vector gene delivery to non-human primates. Gene Ther. 2015, 22, 116–126. [Google Scholar] [CrossRef] [Green Version]
- Fitzpatrick, Z.; Leborgne, C.; Barbon, E.; Masat, E.; Ronzitti, G.; van Wittenberghe, L.; Vignaud, A.; Collaud, F.; Charles, S.; Sola, M.S. Influence of pre-existing anti-capsid neutralizing and binding antibodies on AAV vector transduction. Mol. Ther. Methods Clin. Dev. 2018, 9, 119–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Calcedo, R.; Wang, H.; Bell, P.; Grant, R.; Vandenberghe, L.H.; Sanmiguel, J.; Morizono, H.; Batshaw, M.L.; Wilson, J.M. The pleiotropic effects of natural AAV infections on liver-directed gene transfer in macaques. Mol. Ther. 2010, 18, 126–134. [Google Scholar] [CrossRef]
- Zaiss, A.K.; Cotter, M.J.; White, L.R.; Clark, S.A.; Wong, N.C.W.; Holers, V.M.; Bartlett, J.S.; Muruve, D.A. Complement Is an Essential Component of the Immune Response to Adeno-Associated Virus Vectors. J. Virol. 2008, 82, 2727–2740. [Google Scholar] [CrossRef] [Green Version]
- Denard, J.; Marolleau, B.; Jenny, C.; Rao, T.N.; Fehling, H.J.; Voit, T.; Svinartchouk, F. C-reactive protein (CRP) is essential for efficient systemic transduction of recombinant adeno-associated virus vector 1 (rAAV-1) and rAAV-6 in mice. J. Virol. 2013, 87, 10784–10791. [Google Scholar] [CrossRef] [Green Version]
- Chan, Y.K.; Wang, S.K.; Chu, C.J.; Copland, D.A.; Letizia, A.J.; Costa Verdera, H.; Chiang, J.J.; Sethi, M.; Wang, M.K.; Neidermyer, W.J., Jr.; et al. Engineering adeno-associated viral vectors to evade innate immune and inflammatory responses. Sci. Transl. Med. 2021, 13, eabd3438. [Google Scholar] [CrossRef]
- Short, B.G. Safety evaluation of ocular drug delivery formulations: Techniques and practical considerations. Toxicol. Pathol. 2008, 36, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Rowe-Rendleman, C.L.; Durazo, S.A.; Kompella, U.B.; Rittenhouse, K.D.; Di Polo, A.; Weiner, A.L.; Grossniklaus, H.E.; Naash, M.I.; Lewin, A.S.; Horsager, A.; et al. Drug and Gene Delivery to the Back of the Eye: From Bench to Bedside. Investig. Ophthalmol. Vis. Sci. 2014, 55, 2714–2730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peeters, L.; Sanders, N.N.; Braeckmans, K.; Boussery, K.; Van de Voorde, J.; De Smedt, S.C.; Demeester, J. Vitreous: A Barrier to Nonviral Ocular Gene Therapy. Investig. Ophthalmol. Vis. Sci. 2005, 46, 3553–3561. [Google Scholar] [CrossRef]
- Kim, H.; Robinson, S.B.; Csaky, K.G. Investigating the movement of intravitreal human serum albumin nanoparticles in the vitreous and retina. Pharm. Res. 2009, 26, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Chen, Y.-S.; Green, C.R.; Rupenthal, I.D. Hyaluronic acid coated albumin nanoparticles for targeted peptide delivery in the treatment of retinal ischaemia. Biomaterials 2018, 168, 10–23. [Google Scholar] [CrossRef]
- Mashal, M.; Attia, N.; Martínez-Navarrete, G.; Soto-Sánchez, C.; Fernández, E.; Grijalvo, S.; Eritja, R.; Puras, G.; Pedraz, J.L. Gene delivery to the rat retina by non-viral vectors based on chloroquine-containing cationic niosomes. J. Control. Release 2019, 304, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.; Qiu, S.; Wang, Y.V.; Park, S.J.; Mohns, E.J.; Mehta, B.; Liu, X.; Chang, B.; Zenisek, D.; Crair, M.C. Restoration of vision after de novo genesis of rod photoreceptors in mammalian retinas. Nature 2018, 560, 484–488. [Google Scholar] [CrossRef]
- Devoldere, J.; Peynshaert, K.; De Smedt, S.C.; Remaut, K. Müller cells as a target for retinal therapy. Drug. Discov. 2019, 24, 1483–1498. [Google Scholar] [CrossRef]
- de Jong, S.; Gagliardi, G.; Garanto, A.; de Breuk, A.; Lechanteur, Y.T.E.; Katti, S.; van den Heuvel, L.P.; Volokhina, E.B.; den Hollander, A.I. Implications of genetic variation in the complement system in age-related macular degeneration. Prog. Retin. Eye Res. 2021, 84, 100952. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Huisingh, C.; Messinger, J.; Dolz-Marco, R.; Ferrara, D.; Freund, K.B.; Curcio, C.A. Histology of geographic atrophy secondary to age-related macular degeneration: A multilayer approach. Retina 2018, 38, 1937. [Google Scholar] [CrossRef] [PubMed]
- Scholl, H.P. Complement Inhibition in Age-Related Macular Degeneration—Treat Early! JAMA Ophthalmol. 2022, 140, 250–251. [Google Scholar] [CrossRef]
- Thomas, C.N.; Sim, D.A.; Lee, W.H.; Alfahad, N.; Dick, A.D.; Denniston, A.K.; Hill, L.J. Emerging therapies and their delivery for treating age-related macular degeneration. Br. J. Pharmacol. 2022, 179, 1908–1937. [Google Scholar] [CrossRef]
- Garafalo, A.V.; Cideciyan, A.V.; Héon, E.; Sheplock, R.; Pearson, A.; WeiYang Yu, C.; Sumaroka, A.; Aguirre, G.D.; Jacobson, S.G. Progress in treating inherited retinal diseases: Early subretinal gene therapy clinical trials and candidates for future initiatives. Prog. Retin. Eye Res. 2020, 77, 100827. [Google Scholar] [CrossRef]
- Trapani, I.; Auricchio, A. Seeing the Light after 25 Years of Retinal Gene Therapy. Trends Mol. Med. 2018, 24, 669–681. [Google Scholar] [CrossRef]
- Jüttner, J.; Szabo, A.; Gross-Scherf, B.; Morikawa, R.K.; Rompani, S.B.; Hantz, P.; Szikra, T.; Esposti, F.; Cowan, C.S.; Bharioke, A.; et al. Targeting neuronal and glial cell types with synthetic promoter AAVs in mice, non-human primates and humans. Nat. Neurosci. 2019, 22, 1345–1356. [Google Scholar] [CrossRef]
- Vandenberghe, L.H.; Auricchio, A. Novel adeno-associated viral vectors for retinal gene therapy. Gene Ther. 2012, 19, 162–168. [Google Scholar] [CrossRef] [Green Version]
- Mowat, F.M.; Gornik, K.R.; Dinculescu, A.; Boye, S.L.; Hauswirth, W.W.; Petersen-Jones, S.M.; Bartoe, J.T. Tyrosine capsid-mutant AAV vectors for gene delivery to the canine retina from a subretinal or intravitreal approach. Gene Ther. 2014, 21, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Annamalai, B.; Parsons, N.; Nicholson, C.; Obert, E.; Jones, B.; Rohrer, B. Subretinal Rather Than Intravitreal Adeno-Associated Virus-Mediated Delivery of a Complement Alternative Pathway Inhibitor Is Effective in a Mouse Model of RPE Damage. Investig. Ophthalmol. Vis. Sci. 2021, 62, 11. [Google Scholar] [CrossRef]
- Amado, D.; Mingozzi, F.; Hui, D.; Bennicelli, J.L.; Wei, Z.; Chen, Y.; Bote, E.; Grant, R.L.; Golden, J.A.; Narfstrom, K. Safety and efficacy of subretinal readministration of a viral vector in large animals to treat congenital blindness. Sci. Transl. Med. 2010, 2, 21ra16. [Google Scholar] [CrossRef] [Green Version]
- Ye, G.J.; Budzynski, E.; Sonnentag, P.; Nork, T.M.; Miller, P.E.; Sharma, A.K.; Ver Hoeve, J.N.; Smith, L.M.; Arndt, T.; Calcedo, R.; et al. Safety and Biodistribution Evaluation in Cynomolgus Macaques of rAAV2tYF-PR1.7-hCNGB3, a Recombinant AAV Vector for Treatment of Achromatopsia. Hum. Gene Ther. Clin. Dev. 2016, 27, 37–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandenberghe, L.H.; Bell, P.; Maguire, A.M.; Cearley, C.N.; Xiao, R.; Calcedo, R.; Wang, L.; Castle, M.J.; Maguire, A.C.; Grant, R.; et al. Dosage thresholds for AAV2 and AAV8 photoreceptor gene therapy in monkey. Sci. Transl. Med. 2011, 3, 88ra54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khabou, H.; Cordeau, C.; Pacot, L.; Fisson, S.; Dalkara, D. Dosage Thresholds and Influence of Transgene Cassette in Adeno-Associated Virus-Related Toxicity. Hum. Gene Ther. 2018, 29, 1235–1241. [Google Scholar] [CrossRef]
- MacLachlan, T.K.; Milton, M.N.; Turner, O.; Tukov, F.; Choi, V.W.; Penraat, J.; Delmotte, M.H.; Michaut, L.; Jaffee, B.D.; Bigelow, C.E. Nonclinical Safety Evaluation of scAAV8-RLBP1 for Treatment of RLBP1 Retinitis Pigmentosa. Mol. Ther. Methods Clin. Dev. 2018, 8, 105–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treatment of Leber Congenital Amaurosis Due to RPE65 Mutations by Ocular Subretinal Injection of Adeno-Associated Virus Gene Vector: Short-Term Results of a Phase I Trial. Hum. Gene Ther. 2008, 19, 979–990. [CrossRef] [PubMed] [Green Version]
- Maguire, A.M.; Simonelli, F.; Pierce, E.A.; Pugh, E.N., Jr.; Mingozzi, F.; Bennicelli, J.; Banfi, S.; Marshall, K.A.; Testa, F.; Surace, E.M. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N. Engl. J. Med. 2008, 358, 2240–2248. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, S.G.; Cideciyan, A.V.; Ratnakaram, R.; Heon, E.; Schwartz, S.B.; Roman, A.J.; Peden, M.C.; Aleman, T.S.; Boye, S.L.; Sumaroka, A.; et al. Gene therapy for leber congenital amaurosis caused by RPE65 mutations: Safety and efficacy in 15 children and adults followed up to 3 years. Arch. Ophthalmol. 2012, 130, 9–24. [Google Scholar] [CrossRef] [Green Version]
- Sastry, A.; Li, J.D.; Raynor, W.; Viehland, C.; Song, Z.; Xu, L.; Farsiu, S.; Izatt, J.A.; Toth, C.A.; Vajzovic, L. Microscope-Integrated OCT-Guided Volumetric Measurements of Subretinal Blebs Created by a Suprachoroidal Approach. Transl. Vis. Sci. Technol. 2021, 10, 24. [Google Scholar] [CrossRef]
- Bainbridge, J.W.; Mehat, M.S.; Sundaram, V.; Robbie, S.J.; Barker, S.E.; Ripamonti, C.; Georgiadis, A.; Mowat, F.M.; Beattie, S.G.; Gardner, P.J. Long-term effect of gene therapy on Leber’s congenital amaurosis. N. Engl. J. Med. 2015, 372, 1887–1897. [Google Scholar] [CrossRef] [Green Version]
- Xue, K.; Groppe, M.; Salvetti, A.P.; MacLaren, R.E. Technique of retinal gene therapy: Delivery of viral vector into the subretinal space. Eye 2017, 31, 1308–1316. [Google Scholar] [CrossRef] [Green Version]
- Gardiner, K.L.; Cideciyan, A.V.; Swider, M.; Dufour, V.L.; Sumaroka, A.; Komáromy, A.M.; Hauswirth, W.W.; Iwabe, S.; Jacobson, S.G.; Beltran, W.A. Long-term structural outcomes of late-stage RPE65 gene therapy. Mol. Ther. 2020, 28, 266–278. [Google Scholar] [CrossRef]
- Halász, É.; Townes-Anderson, E.; Zarbin, M.A. Improving outcomes in retinal detachment: The potential role of rho-kinase inhibitors. Curr. Opin. Ophthalmol. 2020, 31, 192–198. [Google Scholar] [CrossRef]
- MacLaren, R.E.; Groppe, M.; Barnard, A.R.; Cottriall, C.L.; Tolmachova, T.; Seymour, L.; Clark, K.R.; During, M.J.; Cremers, F.P.; Black, G.C.; et al. Retinal gene therapy in patients with choroideremia: Initial findings from a phase 1/2 clinical trial. Lancet 2014, 383, 1129–1137. [Google Scholar] [CrossRef] [Green Version]
- Adijanto, J.; Naash, M.I. Nanoparticle-based technologies for retinal gene therapy. Eur. J. Pharm. Biopharm. 2015, 95, 353–367. [Google Scholar] [CrossRef] [Green Version]
- Botto, C.; Rucli, M.; Tekinsoy, M.D.; Pulman, J.; Sahel, J.-A.; Dalkara, D. Early and late stage gene therapy interventions for inherited retinal degenerations. Prog. Retin. Eye Res. 2022, 86, 100975. [Google Scholar] [CrossRef]
- Einmahl, S.; Savoldelli, M.; D’Hermies, F.o.; Tabatabay, C.; Gurny, R.; Behar-Cohen, F. Evaluation of a novel biomaterial in the suprachoroidal space of the rabbit eye. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1533–1539. [Google Scholar]
- Chiang, B.; Kim, Y.C.; Edelhauser, H.F.; Prausnitz, M.R. Circumferential flow of particles in the suprachoroidal space is impeded by the posterior ciliary arteries. Exp. Eye Res. 2016, 145, 424–431. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.R.; Lin, A.S.; Edelhauser, H.F.; Prausnitz, M.R. Suprachoroidal drug delivery to the back of the eye using hollow microneedles. Pharm. Res. 2011, 28, 166–176. [Google Scholar] [CrossRef]
- Olsen, T.W.; Feng, X.; Wabner, K.; Csaky, K.; Pambuccian, S.; Cameron, J.D. Pharmacokinetics of pars plana intravitreal injections versus microcannula suprachoroidal injections of bevacizumab in a porcine model. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4749–4756. [Google Scholar] [CrossRef] [Green Version]
- Rai, U.D.J.P.; Young, S.A.; Thrimawithana, T.R.; Abdelkader, H.; Alani, A.W.G.; Pierscionek, B.; Alany, R.G. The suprachoroidal pathway: A new drug delivery route to the back of the eye. Drug. Discov. 2015, 20, 491–495. [Google Scholar] [CrossRef]
- Peden, M.C.; Min, J.; Meyers, C.; Lukowski, Z.; Li, Q.; Boye, S.L.; Levine, M.; Hauswirth, W.W.; Ratnakaram, R.; Dawson, W. Ab-externo AAV-mediated gene delivery to the suprachoroidal space using a 250 micron flexible microcatheter. PLoS ONE 2011, 6, e17140. [Google Scholar] [CrossRef] [Green Version]
- Chiang, B.; Venugopal, N.; Edelhauser, H.F.; Prausnitz, M.R. Distribution of particles, small molecules and polymeric formulation excipients in the suprachoroidal space after microneedle injection. Exp. Eye Res. 2016, 153, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.H.; Mollhoff, I.N.; Mishra, A.; Sin, T.-N.; Ngo, T.; Ciulla, T.; Sieving, P.; Thomasy, S.M.; Yiu, G. Host immune responses after suprachoroidal delivery of AAV8 in nonhuman primate eyes. Hum. Gene Ther. 2021, 32, 682–693. [Google Scholar] [CrossRef]
- Ding, K.; Shen, J.; Hafiz, Z.; Hackett, S.F.; e Silva, R.L.; Khan, M.; Lorenc, V.E.; Chen, D.; Chadha, R.; Zhang, M. AAV8-vectored suprachoroidal gene transfer produces widespread ocular transgene expression. J. Clin. Investig. 2019, 129, 4901–4911. [Google Scholar] [CrossRef]
- Yiu, G.; Chung, S.H.; Mollhoff, I.N.; Nguyen, U.T.; Thomasy, S.M.; Yoo, J.; Taraborelli, D.; Noronha, G. Suprachoroidal and Subretinal Injections of AAV Using Transscleral Microneedles for Retinal Gene Delivery in Nonhuman Primates. Mol. Ther. Methods Clin. Dev. 2020, 16, 179–191. [Google Scholar] [CrossRef] [Green Version]
- Kansara, V.; Muya, L.; Wan, C.-r.; Ciulla, T.A. Suprachoroidal delivery of viral and nonviral gene therapy for retinal diseases. J. Ocul. Pharmacol. Ther. 2020, 36, 384–392. [Google Scholar] [CrossRef]
- Hancock, S.E.; Phadke, A.; Kansara, V.; Boyer, D.; Rivera, J.; Marlor, C.; Podos, S.; Wiles, J.; McElheny, R.; Ciulla, T.A.; et al. Ocular Pharmacokinetics and Safety of Suprachoroidal A01017, Small Molecule Complement Inhibitor, Injectable Suspension in Rabbits. Investig. Ophthalmol. Vis. Sci. 2020, 61, 3694. [Google Scholar]






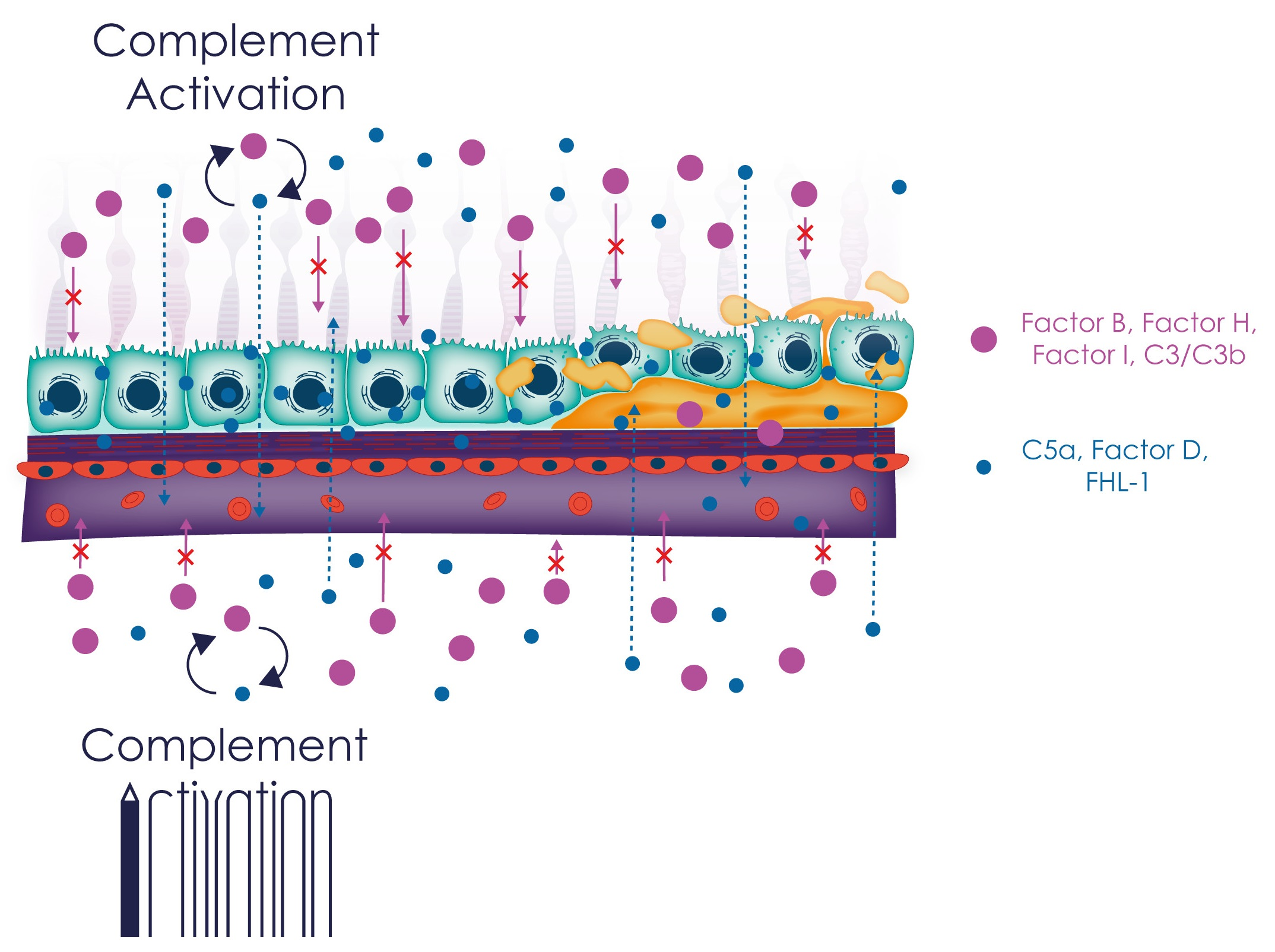{kind=link}
{kind=link}
{kind=link}
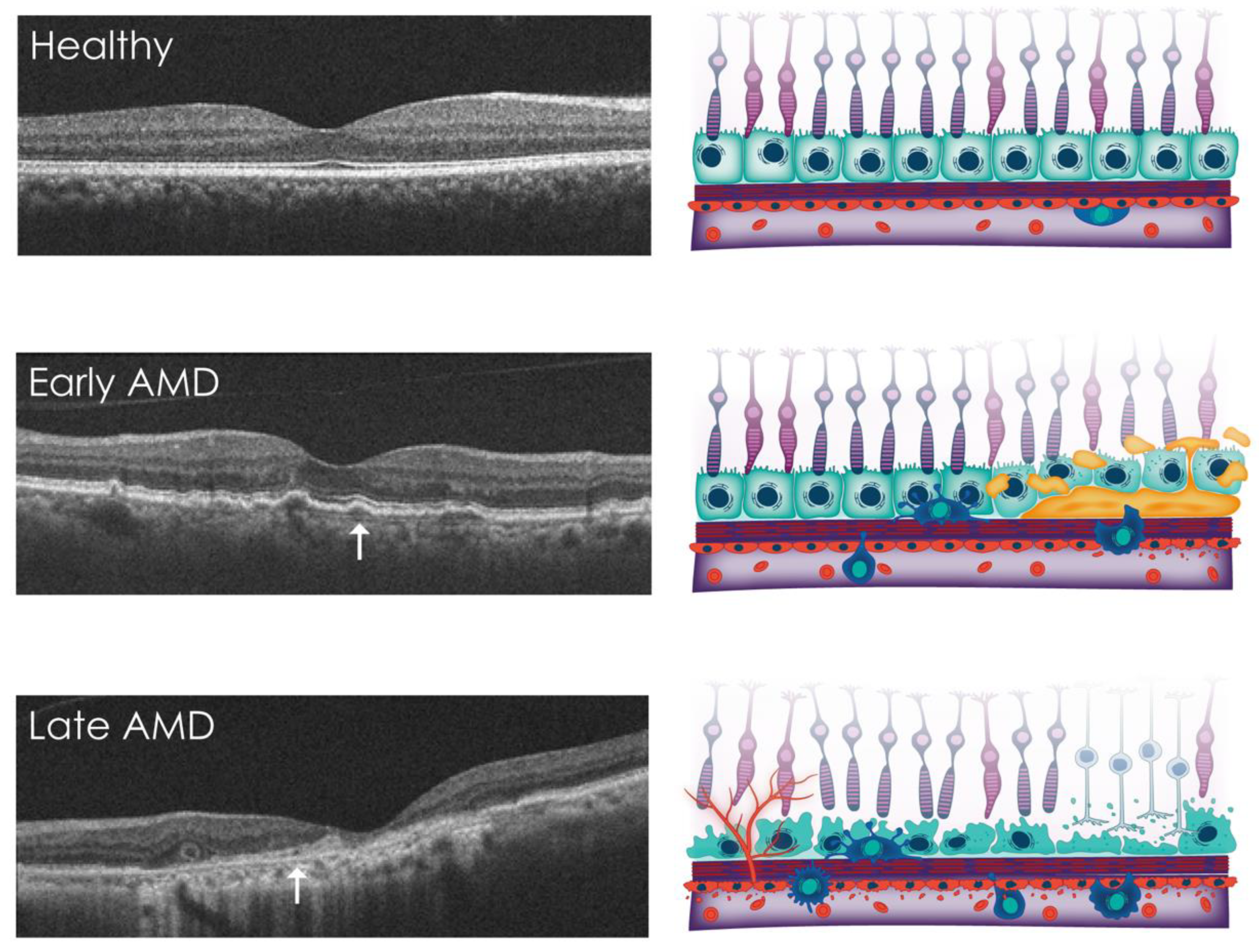{kind=link}
{kind=link}
{kind=link}
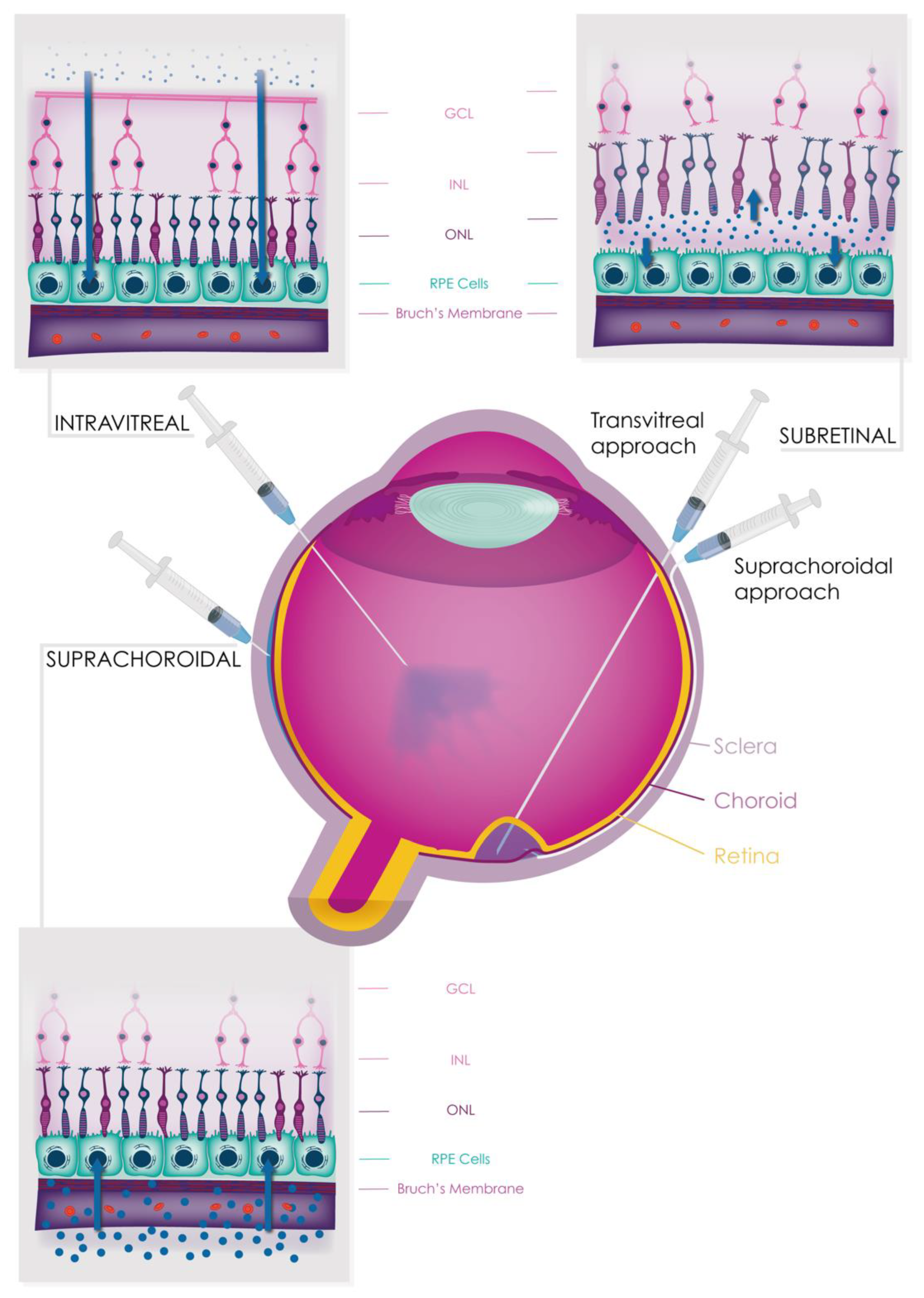{kind=link}
| Functional Changes | Anatomical Changes |
|---|---|
| Decrease in elasticity Decrease in water permeability Decrease in protein permeability Decreased complement protein permeability | Accumulation of lipids, TIMP-3, MMP-2, MMP-9, calcium phosphate, and AGEs BrM thickening Reduced heparan sulphate Increased complement activation |
| Layers | Composition | Changes with Age |
|---|---|---|
| RPE basement membrane | Chondroitin sulphate Collagen IV α1–5 Collagen V Heparan sulphate Laminins 1, 5, 10, and 11 Nidogen-1 | BLamD accumulation |
| Inner collagenous layer | Apolipoprotein E Chondroitin sulphate Clusterin Collagen I, III, and V Dermatan sulphate Fibronectin Haem Lipoproteins Vitronectin | Lipoprotein deposition BLinD accumulation |
| Central elastic layer | Elastin | Elastin and calcium phosphate deposition |
| Outer collagenous layer | Apolipoprotein E Chondroitin sulphate Clusterin Collagen I, III, and V Dermatan sulphate Fibronectin Fibulin-5 Lipoproteins | Lipoprotein deposition |
| Choroidal endothelial cell basement membrane | Chondroitin sulphate Collagen IV, α1, α2 Collagen V Collagen VI Endostatin Heparan sulphate Laminin | Unknown |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hammadi, S.; Tzoumas, N.; Ferrara, M.; Meschede, I.P.; Lo, K.; Harris, C.; Lako, M.; Steel, D.H. Bruch’s Membrane: A Key Consideration with Complement-Based Therapies for Age-Related Macular Degeneration. J. Clin. Med. 2023, 12, 2870. https://doi.org/10.3390/jcm12082870
Hammadi S, Tzoumas N, Ferrara M, Meschede IP, Lo K, Harris C, Lako M, Steel DH. Bruch’s Membrane: A Key Consideration with Complement-Based Therapies for Age-Related Macular Degeneration. Journal of Clinical Medicine. 2023; 12(8):2870. https://doi.org/10.3390/jcm12082870
Chicago/Turabian StyleHammadi, Sarah, Nikolaos Tzoumas, Mariantonia Ferrara, Ingrid Porpino Meschede, Katharina Lo, Claire Harris, Majlinda Lako, and David H. Steel. 2023. "Bruch’s Membrane: A Key Consideration with Complement-Based Therapies for Age-Related Macular Degeneration" Journal of Clinical Medicine 12, no. 8: 2870. https://doi.org/10.3390/jcm12082870