
Haplotype-Based Single-Step GWAS for Yearling Temperament in American Angus Cattle



Abstract
:1. Introduction
2. Materials and Methods
2.1. Phenotypic and Pedigree Data
2.2. Genotypic Data
2.3. Haplotype Block Construction
2.4. Single-Step GWAS with Haplotypes
2.5. Weighted Single-Step GWAS with Haplotypes
2.6. Scenarios Evaluated
2.7. Empirical Selection of the Candidate Regions for Further Investigation
2.8. Functional Analyses
3. Results
3.1. Statistics from Haplotype Blocking
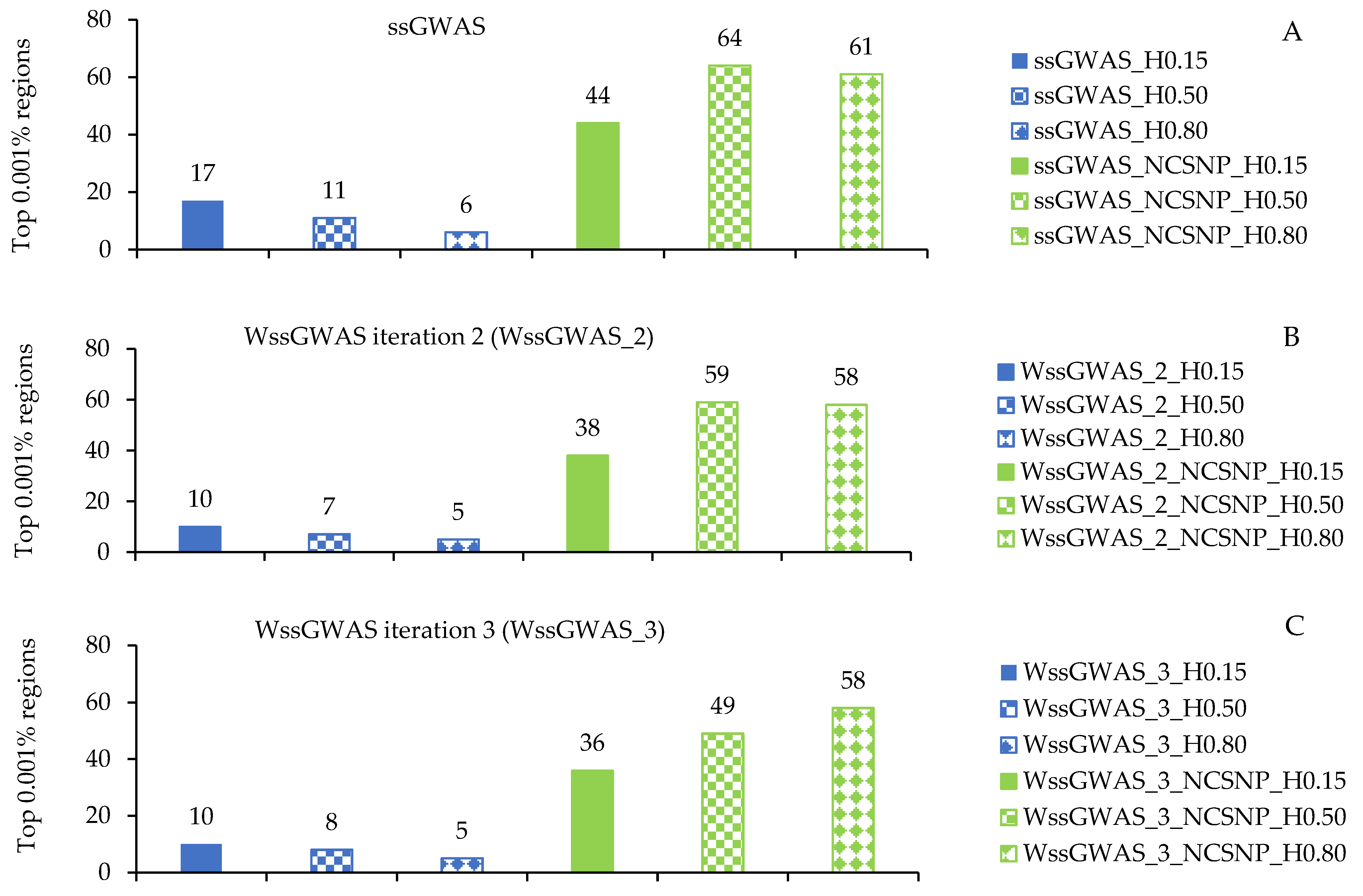
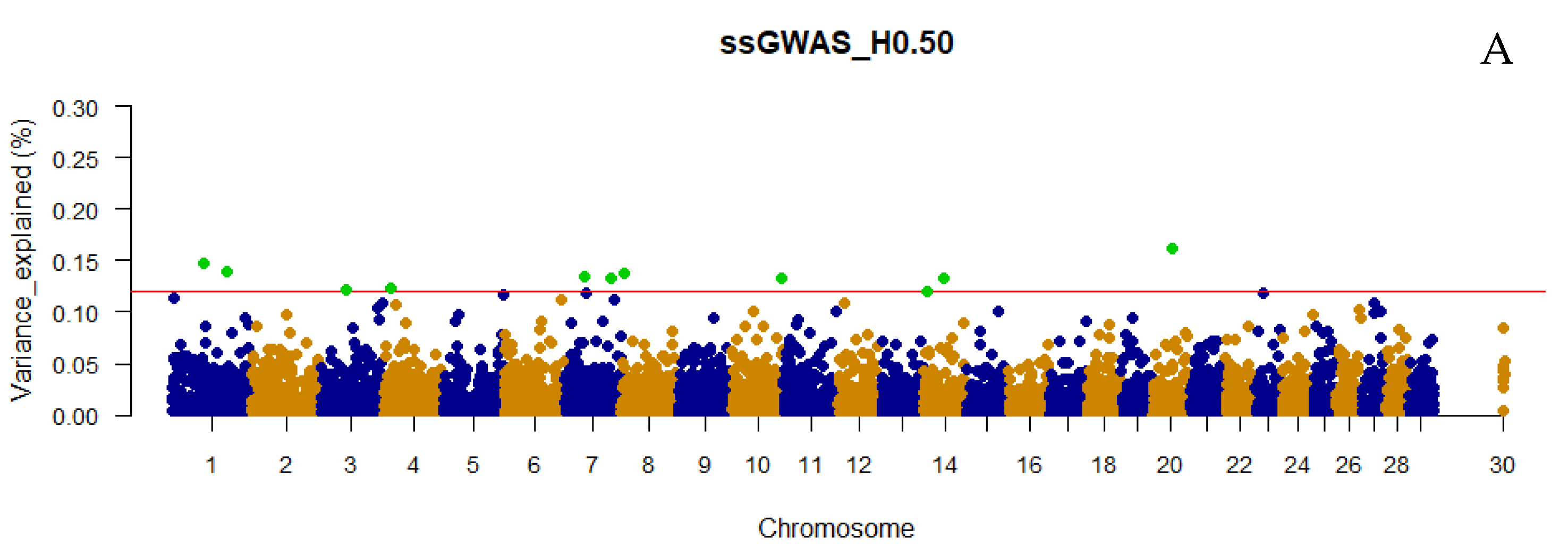
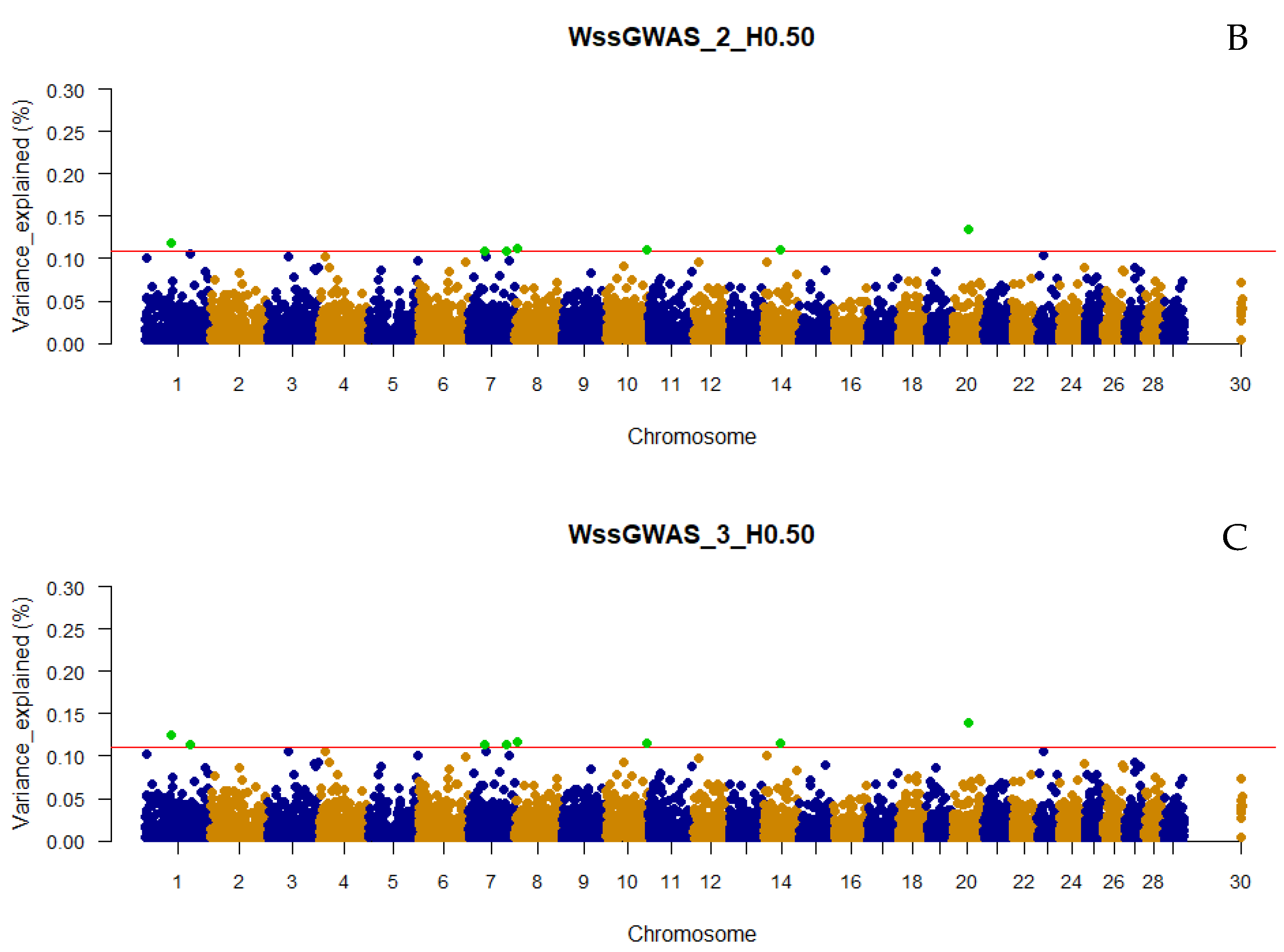
3.2. Traditional and Weighted Single-Step GWAS Fitting Only Haplotypes
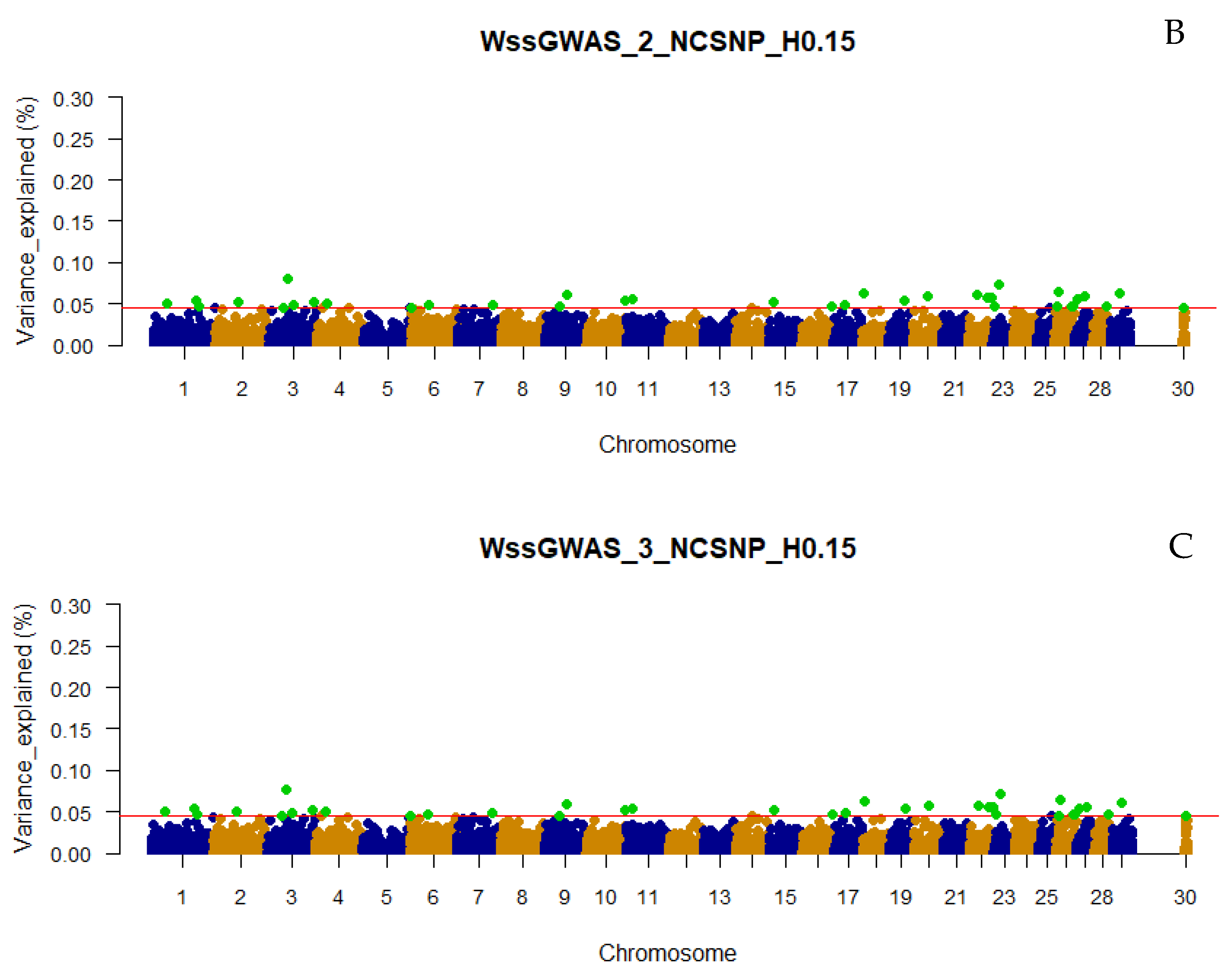
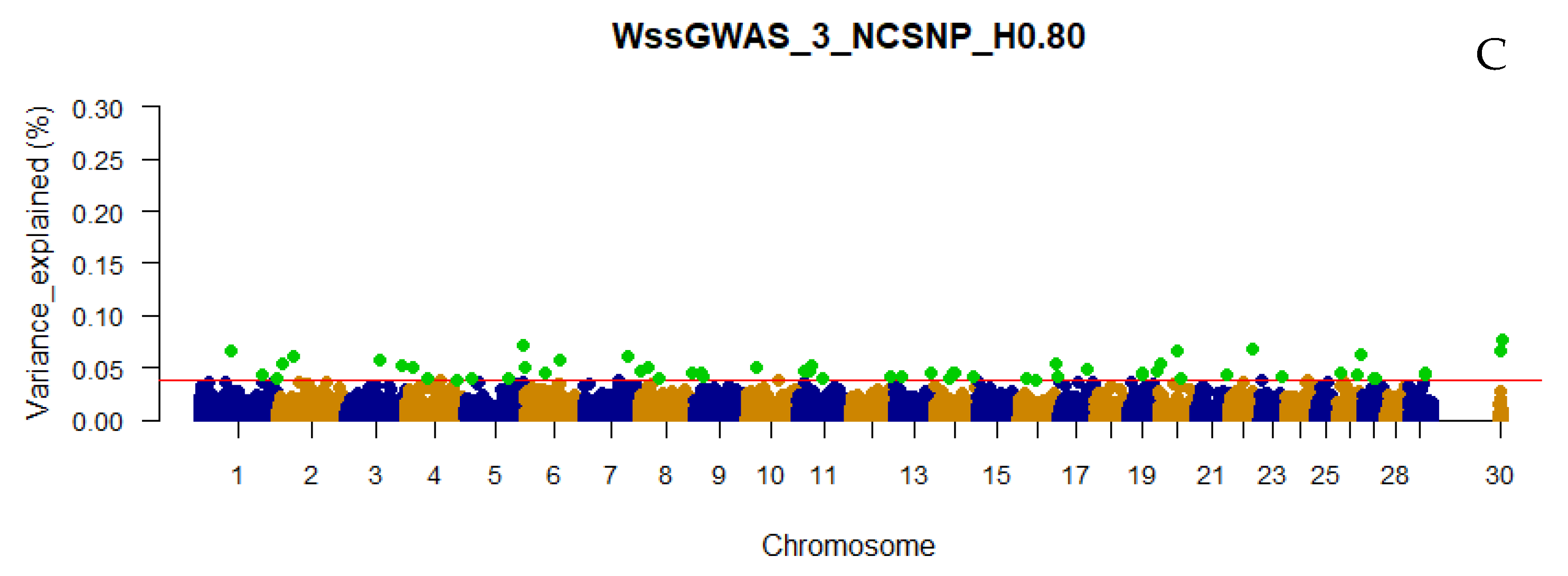
3.3. Traditional and Weighted Single-Step GWAS Fitting Haplotype Blocks and Non-Clustered SNP
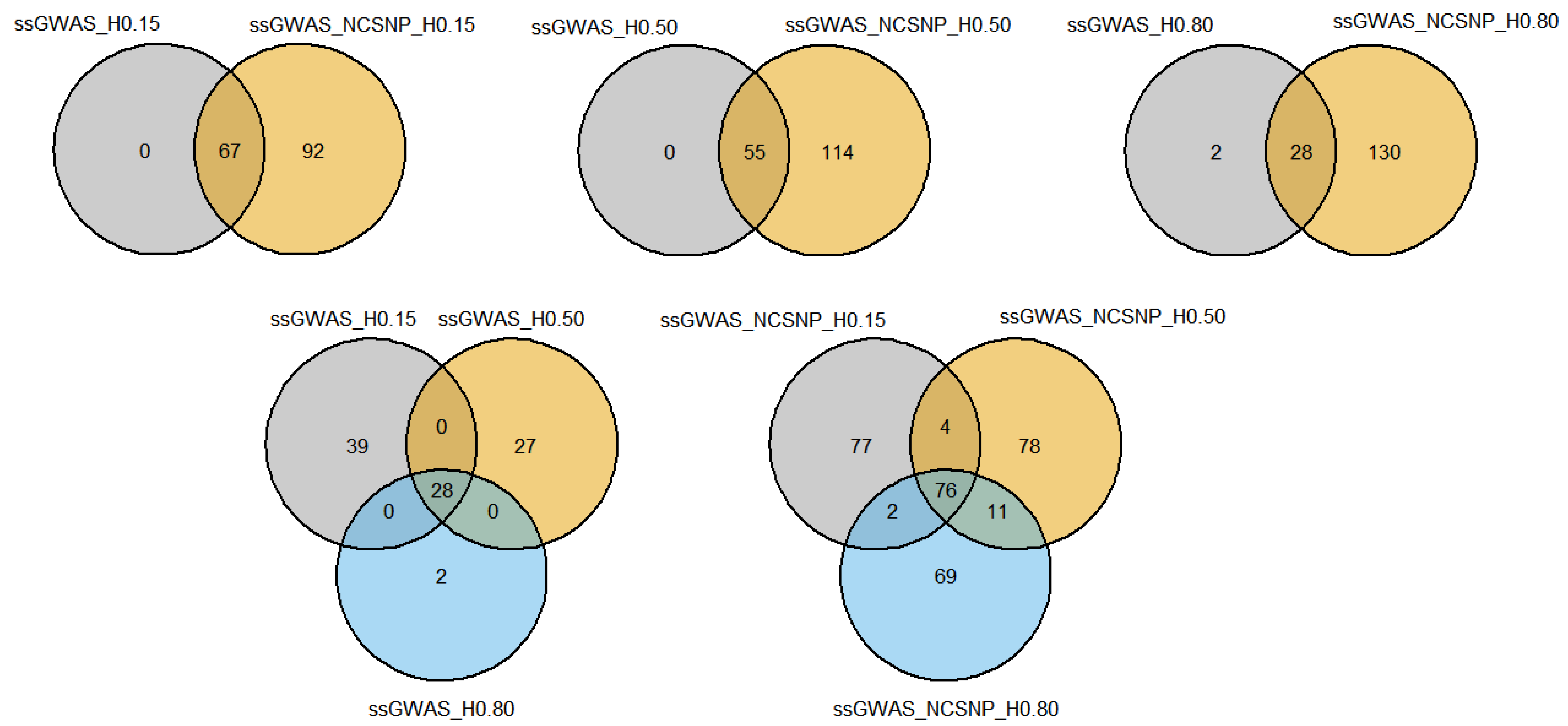
3.4. Overlapping Genomic Regions among Methods and Functional Analyses
3.4.1. Overlapping Markers
3.4.2. Overlapping Genes
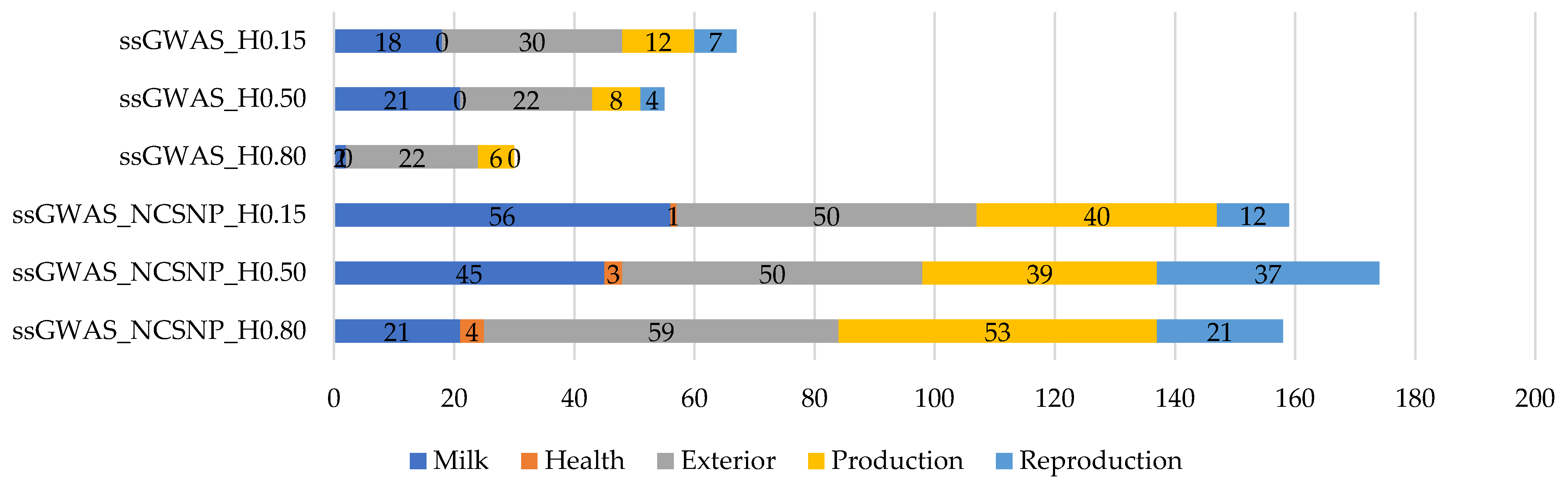
3.4.3. Functional Analyses
3.5. QTL Overlapping with the Top 0.001% Markers for Yearling Temperament
4. Discussion
4.1. Empirical Selection of the Candidate Genomic Regions
4.2. Additive Genetic Variance Explainded by Genomic Regions across Scenarios
4.3. Weighting Method in the Single-Step GWAS
4.4. Genes and QTL Overlapping the Top Genomic Regions
4.5. Use of Different Linkage Disequilibrium Thresolds and Non-Clustered SNPs in the ssGWAS
4.6. Future Studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grandin, T.; Deesing, M.J. Behavioral genetics and animal science. In Genetics and the Behavior of Domestic Animals, 2nd ed.; Academic Press: San Diego, CA, USA, 2014; pp. 1–40. [Google Scholar]
- Steimer, T. The biology of fear- and anxiety-related behaviors. Dialogues Clin. Neurosci. 2002, 4, 231–249. [Google Scholar]
- Valente, T.S.; Baldi, F.; Sant’Anna, A.C.; Albuquerque, L.G.; Paranhos da Costa, M.J. Genome-wide association study between single nucleotide polymorphisms and flight speed in Nellore Cattle. PLoS ONE 2016, 11, e0156956. [Google Scholar] [CrossRef]
- Costilla, R.; Kemper, K.E.; Byrne, E.M.; Porto-Neto, L.R.; Carvalheiro, R.; Purfield, D.C.; Doyle, J.L.; Berry, D.P.; Moore, S.S.; Wray, N.R.; et al. Genetic control of temperament traits across species: Association of autism spectrum disorder risk genes with cattle temperament. Genet. Sel. Evol. 2020, 52, 1–14. [Google Scholar] [CrossRef]
- Alvarenga, A.B.; Oliveira, H.R.; Miller, S.P.; Silva, F.F.; Brito, L.F. Genetic modeling and genomic analysis of yearling temperament in American Angus Cattle and its relationship with productive efficiency and resilience traits. Front. Genet. under review.
- Cooke, R.F.; Moriel, P.; Cappellozza, B.I.; Miranda, V.F.B.; Batista, L.F.D.; Colombo, E.A.; Ferreira, V.S.M.; Miranda, M.F.; Marques, R.S.; Vasconcelos, J.L.M. Effects of temperament on growth, plasma cortisol concentrations and puberty attainment in Nelore beef heifers. Animal 2019, 13, 1208–1213. [Google Scholar] [CrossRef]
- By the Numbers: Docility Genetic Evaluation Research. Available online: http://www.angus.org/nce/documents/bythenumbersdocility.pdf (accessed on 12 August 2021).
- Alvarenga, A.B.; Oliveira, H.R.; Chen, S.Y.; Miller, S.P.; Marchant-Forde, J.N.; Grigoletto, L.; Brito, L.F. A systematic review of genomic regions and candidate genes underlying behavioral traits in farmed mammals and their link with human disorders. Animals 2021, 11, 715. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, S.B.; Schaffner, S.F.; Nguyen, H.; Moore, J.M.; Roy, J.; Blumenstiel, B.; Higgins, J.; DeFelice, M.; Lochner, A.; Faggart, M.; et al. The structure of haplotype blocks in the human genome. Science 2002, 296, 2225–2229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calus, M.P.L.; Meuwissen, T.H.E.; de Roos, A.P.W.; Veerkamp, R.F. Accuracy of genomic selection using different methods to define haplotypes. Genetics 2008, 178, 553–561. [Google Scholar] [CrossRef] [Green Version]
- Villumsen, T.M.; Janss, L.; Lund, M.S. The importance of haplotype length and heritability using genomic selection in dairy cattle. J. Anim. Breed. Genet. 2009, 126, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Hess, M.; Druet, T.; Hess, A.; Garrick, D. Fixed-length haplotypes can improve genomic prediction accuracy in an admixed dairy cattle population. Genet. Sel. Evol. 2017, 49, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Schmidt, R.H.; Reif, J.C. Haplotype-based genome-wide prediction models exploit local epistatic interactions among markers. G3 2018, 8, 1687–1699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Z.; Tan, C.; Prakapenka, D.; Ma, L.; Da, Y. Haplotype analysis of genomic prediction using structural and functional genomic information for seven human phenotypes. Front. Genet. 2020, 11, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Braz, C.U.; Taylor, J.F.; Bresolin, T.; Espigolan, R.; Feitosa, F.L.B.; Carvalheiro, R.; Baldi, F.; Albuquerque, L.G.; Oliveira, H.N. Sliding window haplotype approaches overcome single SNP analysis limitations in identifying genes for meat tenderness in Nelore cattle. BMC Genet. 2019, 20, 1–12. [Google Scholar] [CrossRef]
- Bovo, S.; Ballan, M.; Schiavo, G.; Ribani, A.; Tinarelli, S.; Utzeri, V.J.; Dall’Olio, S.; Gallo, M.; Fontanesi, L. Single-marker and haplotype-based genome-wide association studies for the number of teats in two heavy pig breeds. Anim. Genet. 2021, 52, 440–450. [Google Scholar] [CrossRef]
- Martin, E.R.; Lai, E.H.; Gilbert, J.R.; Rogala, A.R.; Afshari, A.J.; Riley, J.; Finch, K.L.; Stevens, J.F.; Livak, K.J.; Slotterbeck, B.D.; et al. SNPing away at complex diseases: Analysis of single-nucleotide polymorphisms around APOE in Alzheimer disease. Am. J. Hum. Genet. 2000, 67, 383–394. [Google Scholar] [CrossRef] [Green Version]
- Araujo, A.C.; Carneiro, P.L.S.; Oliveira, H.R.; Schenkel, F.S.; Veroneze, R.; Lourenco, D.A.L.; Brito, L.F. A comprehensive comparison of haplotype-based single-step genomic predictions in livestock populations with different genetic diversity levels: A simulation study. Front. Genet. 2021, 12, 1–17. [Google Scholar]
- Wang, H.; Misztal, I.; Aguilar, I.; Legarra, A.; Muir, W.M. Genome-wide association mapping including phenotypes from relatives without genotypes. Genet. Res. 2012, 94, 73–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancin, E.; Lourenco, D.; Bermann, M.; Mantovani, R.; Misztal, I. Accounting for population structure and phenotypes from relatives in association mapping for farm animals: A simulation study. Front. Genet. 2021, 12, 1–14. [Google Scholar]
- Legarra, A.; Aguilar, I.; Misztal, I. A Relationship Matrix Including Full Pedigree and Genomic Information. J. Dairy Sci. 2009, 92, 4656–4663. [Google Scholar] [CrossRef] [Green Version]
- Aguilar, I.; Misztal, I.; Johnson, D.L.; Legarra, A.; Tsuruta, S.; Lawlor, T.J. Hot topic: A unified approach to utilize phenotypic, full pedigree, and genomic information for genetic evaluation of Holstein final score. J. Dairy Sci. 2010, 93, 743–752. [Google Scholar] [CrossRef]
- Zhang, X.; Lourenco, D.; Aguilar, I.; Legarra, A.; Misztal, I. Weighting strategies for single-step genomic BLUP: An iterative approach for accurate calculation of GEBV and GWAS. Front. Genet. 2016, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Johnson, T.; Keehan, M.; Harland, C.; Lopdell, T.; Spelman, R.J.; Davis, S.R.; Rosen, B.D.; Smith, T.P.L.; Couldrey, C. Short communication: Identification of the pseudoautosomal region in the Hereford bovine reference genome assembly ARS-UCD1.2. J. Dairy Sci. 2019, 102, 3254–3258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misztal, I.; Tsuruta, S.; Lourenco, D.A.L.; Masuda, Y.; Aguilar, I.; Legarra, A.; Vitezica, Z. Manual for BLUPF90 Family Programs; University of Georgia: Athens, GA, USA, 2018; Available online: http://nce.ads.uga.edu/wiki/lib/exe/fetch.php?media=blupf90_all2.pdf (accessed on 12 June 2021).
- Sargolzaei, M.; Chesnais, J.P.; Schenkel, F.S. A new approach for efficient genotype imputation using information from relatives. BMC Genom. 2014, 15, 478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, W.G.; Robertson, A. Linkage disequilibrium in finite populations. Theoret. Appl. Genet. 1968, 38, 226–231. [Google Scholar] [CrossRef]
- Kim, S.A.; Cho, C.-S.; Kim, S.-R.; Bull, S.B.; Yoo, Y.J. A new haplotype block detection method for dense genome sequencing data based on interval graph modeling of clusters of highly correlated SNPs. Bioinformatics 2018, 34, 388–397. [Google Scholar] [CrossRef]
- Kim, S.A.; Brossard, M.; Roshandel, D.; Paterson, A.D.; Bull, S.B.; Yoo, Y.J. gpart: Human genome partitioning and visualization of high-density SNP data by identifying haplotype blocks. Bioinformatics 2019, 35, 4419–4421. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation For Statistical Computing: Vienna, Austria, 2020; Available online: www.R-project.org/ (accessed on 10 June 2021).
- Teissier, M.; Larroque, H.; Brito, L.F.; Rupp, R.; Schenkel, F.S.; Robert-Granié, C. Genomic predictions based on haplotypes fitted as pseudo-SNP for milk production and udder type traits and SCS in French dairy goats. J. Dairy Sci. 2020, 103, 11559–11573. [Google Scholar] [CrossRef]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [Green Version]
- Strandén, I.; Garrick, D.J. Derivation of equivalent computing algorithms for genomic predictions and reliabilities of animal merit. J. Dairy Sci. 2009, 92, 2971–2975. [Google Scholar] [CrossRef] [Green Version]
- Fragomeni, B.O.; Lourenco, D.A.L.; Legarra, A.; VanRaden, P.; Misztal, I. Alternative SNP weighting for single-step genomic best linear unbiased predictor evaluation of stature in US Holsteins in the presence of selected sequence variants. J. Dairy Sci. 2019, 102, 10012–10019. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sung, W.K.; Liu, J.J. Association mapping via regularized regression analysis of single-nucleotide polymorphism haplotypes in variable-sized sliding windows. Am. J. Hum. Genet. 2007, 80, 705–715. [Google Scholar] [CrossRef] [Green Version]
- Cullen, A.; Frey, H. Probabilistic Techniques in Exposure Assessment, 1st ed.; Plenum Publishing Co: New York, NY, USA; Springer: New York, NY, USA, 1999. [Google Scholar]
- Delignette-Muller, M.L.; Dutang, C. fitdistrplus: An R Package for Fitting Distributions. J. Stat. Softw. 2015, 64, 1–34. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.L.; Park, C.A.; Reecy, J.M. Building a livestock genetic and genomic information knowledgebase through integrative developments of Animal QTLdb and CorrDB. Nucleic Acids Res. 2019, 47, D701–D710. [Google Scholar] [CrossRef] [Green Version]
- Medrano, J.F. The new bovine reference assembly and its value for genomic research. Proc. Assoc. Advmt. Anim. Breed. Genet. 2017, 22, 161–166. [Google Scholar]
- Rosen, B.D.; Bickhart, D.M.; Schnabel, R.D.; Koren, S.; Elsik, C.G.; Zimin, A.; Dreischer, C.; Schultheiss, S.; Hall, R.; Schroeder, S.G.; et al. Modernizing the bovine reference genome assembly. Proc. World Congr. Genet. Appl. Livest Prod. 2018, 3, 802. [Google Scholar]
- Aguilar, I.; Legarra, A.; Cardoso, F.; Masuda, Y.; Lourenco, D.; Misztal, I. Frequentist p-values for large-scale-single step genome-wide association, with an application to birth weight in American Angus cattle. Genet. Sel. Evol. 2019, 51, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.Y.; Oliveira, H.O.; Schenkel, F.S.; Pedrosa, V.B.; Melka, M.G.; Brito, L.F. Using imputed whole-genome sequence variants to uncover candidate mutations and genes affecting milking speed and temperament in Holstein cattle. J. Dairy Sci. 2020, 103, 10383–10398. [Google Scholar] [CrossRef]
- Mehrban, H.; Naserkheil, M.; Lee, D.H.; Cho, C.; Choi, T.; Park, M.; Ibáñez-Escriche, N. Genomic prediction using alternative strategies of weighted single-step genomic BLUP for yearling weight and carcass traits in Hanwoo beef cattle. Genes 2021, 12, 266. [Google Scholar] [CrossRef] [PubMed]
- Suchocki, T.; Szyda, J. Genome-wide association study for semen production traits in Holstein-Friesian bulls. J. Dairy Sci. 2015, 98, 5774–5780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, D.G.; Gill, C.A.; Boldt, C.R.; Funkhouser, R.R.; Herring, A.D.; Riggs, P.K.; Sawyer, J.E.; Lunt, D.K.; Sanders, J.O. Crossbred Bos indicus steer temperament as yearlings and whole genome association of steer temperament as yearlings and calf temperament post-weaning. J. Anim. Sci. 2016, 94, 1408–1414. [Google Scholar] [CrossRef] [Green Version]
- Michenet, A.; Saintilan, R.; Venot, E.; Phocas, F. Insights into the genetic variation of maternal 1187 behavior and suckling performance of continental beef cows. Genet. Sel. Evol. 2016, 48, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Dreher, C.; Wellmann, R.; Stratz, P.; Schmid, M.; Preuß, S.; Hamann, H.; Bennewitz, J. Genomic analysis of perinatal sucking reflex in German Brown Swiss calves. J. Dairy Sci. 2019, 102, 6296–6305. [Google Scholar] [CrossRef]
- Eddy, S.R. Non-coding RNA genes and the modern RNA world. Nat. Rev. Genet. 2001, 2, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Taye, M.; Lee, W.; Jeo, S.; Yoon, J.; Dessie, T.; Hanotte, O.; Mwai, O.A.; Kemp, S.; Cho, S.; Oh, S.J.; et al. Exploring evidence of positive selection signatures in cattle breeds selected for different traits. Mamm. Genome 2017, 28, 528–541. [Google Scholar] [CrossRef] [PubMed]
- Brito, L.F.; Oliveira, H.R.; McConn, B.R.; Schinckel, A.P.; Arrazola, A.; Marchant-Forde, J.N.; Johnson, J.S. Large-scale phenotyping of livestock welfare in commercial production systems: A new frontier in animal breeding. Front. Genet. 2020, 11, 793. [Google Scholar] [CrossRef]
- Cheng, H.W. Breeding of tomorrow’s chickens to improve well-being. Poult. Sci. 2010, 89, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, A.; Wang, Y.; Luo, H.; Yan, X.; Guo, X.; Li, X.; Liu, L.; Su, G. Genetic parameters and genome-wide association studies of eight longevity traits representing either full or partial lifespan in Chinese Holsteins. Front. Genet. 2021, 12, 634986. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, H.R.; Brito, L.F.; Miller, S.P.; Schenkel, F.S. Using random regression models to genetically evaluate functional longevity traits in North American angus cattle. Animals 2020, 10, 2410. [Google Scholar] [CrossRef]
- Pereira Vatanabe, I.; Peron, R.; Mantellatto Grigoli, M.; Pelucchi, S.; De Cesare, G.; Magalhães, T.; Manzine, P.R.; Figueredo Balthazar, M.L.; Di Luca, M.; Marcello, E.; et al. ADAM10 plasma and CSF levels are increased in mild Alzheimer’s disease. Int. J. Mol. Sci. 2021, 22, 2416. [Google Scholar] [CrossRef] [PubMed]
- Sollero, B.P.; Junqueira, V.S.; Gomes, C.C.G.; Caetano, A.R.; Cardoso, F.F. Tag SNP selection for prediction of tick resistance in Brazilian Braford and Hereford cattle breeds using Bayesian methods. Genet. Sel. Evol. 2017, 49, 49. [Google Scholar] [CrossRef] [Green Version]
- Kasarapu, P.; Porto-Neto, L.R.; Fortes, M.R.S.; Lehnert, S.A.; Mudadu, M.A.; Coutinho, L.; Regitano, L.; George, A.; Reverter, A. The Bos taurus-Bos indicus balance in fertility and milk related genes. PLoS ONE 2017, 12, e0181930. [Google Scholar] [CrossRef]
- Silva, R.P.; Berton, M.P.; Grigoletto, L.; Carvalho, F.E.; Silva, R.M.O.; Peripolli, E.; Castro, L.M.; Ferraz, J.B.S.; Eler, J.P.; Lobo, R.B.; et al. Genomic regions and enrichment analyses associated with carcass composition indicator traits in Nellore cattle. J. Anim. Breed. Genet. 2018, 136, 1–16. [Google Scholar] [CrossRef]
- LaMantia, A.-S. Why does the face predict the brain? Neural crest induction, craniofacial morphogenesis, and neural circuit development. Front. Physiol. 2020, 11, 610970. [Google Scholar] [CrossRef]
- Carre, J.M.; McCormick, C.M.; Mondloch, C.J. Facial structure is a reliable cue of aggressive behavior. Psychol. Sci. 2009, 20, 1194–1198. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Sun, D.; Zhang, S.; Wang, S.; Wu, X.; Zhang, Q.; Liu, L.; Li, Y.; Qiao, L. Genome wide association study identifies 20 novel promising genes associated with milk fatty acid traits in Chinese Holstein. PLoS ONE 2014, 9, e96186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grandin, T.; Deesing, M.J.; Struthers, J.J.; Swinker, A.M. Cattle with hair whorl patterns above the eyes are more behaviorally agitated during restraint. Appl. Anim. Behav. Sci. 1995, 46, 117–123. [Google Scholar] [CrossRef]
- Lanier, J.L.; Grandin, T.; Green, R.D.; Avery, D.; Mcgee, K. Cattle hair whorl position and temperament in auction houses. J. Anim. Sci. 1999, 77, 147. [Google Scholar]
- Lanier, J.L.; Grandin, T.; Green, R.D.; Avery, D.; Mcgee, K. A note on hair whorl position and cattle temperament in the auction ring. Appl. Anim. Behav. Sci. 2001, 73, 93–101. [Google Scholar] [CrossRef]
- Furdon, S.A.; Clark, D.A. Scalp hair characteristics in the newborn infant. Adv. Neonatal Care 2003, 3, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Lima, D.F.P.d.A.; da Cruz, V.A.R.; Pereira, G.L.; Curi, R.A.; Costa, R.B.; de Camargo, G.M.F. Genomic Regions Associated with the Position and Number of Hair Whorls in Horses. Animals 2021, 11, 2925. [Google Scholar] [CrossRef]
- Anilkumar, S.; Patel, D.; Boer, S.F.; Chattarji, S.; Buwalda, B. Decreased dendritic spine density in poster dorsal medial amygdala neurons of proactive coping rats. Behav. Brain Res. 2021, 397, 112940. [Google Scholar] [CrossRef]
- Neupane, M.; Kiser, J.N.; The Bovine Respiratory Disease Complex Coordinated Agricultural Project Research Team; Neibergs, H.L. Gene set enrichment analysis of SNP data in dairy and beef cattle with bovine respiratory disease. Anim. Genet. 2018, 49, 527–538. [Google Scholar] [CrossRef]
- Hay, E.L.; Roberts, A. Genome-wide association study for carcass traits in a composite beef cattle breed. Livest. Sci. 2018, 213, 35–43. [Google Scholar] [CrossRef]
- Bonnefil, V.; Dietz, K.; Amatruda, M.; Wentling, M.; Aubry, A.V.; Dupree, J.L.; Temple, G.; Park, H.J.; Burghardt, N.S.; Casaccia, P. Region-specific myelin differences define behavioral consequences of chronic social defeat stress in mice. eLife 2019, 8, e40855. [Google Scholar] [CrossRef] [PubMed]
- Hartline, D.K. What is myelin? Neuron Glia Biol. 2008, 4, 153–163. [Google Scholar] [CrossRef] [Green Version]
- Huson, H.J.; Kim, E.S.; Godfrey, R.W.; Olson, T.A.; McClure, M.C.; Chase, C.C.; Rizzi, R.; O’Brien, A.M.P.; VanTassell, C.P.; Garcia, J.F. Genome-wide association study and ancestral origins of the slick-hair coat in tropically adapted cattle. Front. Genet. 2014, 5, 1–12. [Google Scholar] [CrossRef]
- Sweett, H.; Fonseca, P.A.S.; Suárez-Vega, A.; Livernois, A.; Miglior, F.; Cánovas, A. Genome-wide association study to identify genomic regions and positional candidate genes associated with male fertility in beef cattle. Sci. Rep. 2020, 10, 20102. [Google Scholar] [CrossRef]
- Manuck, S.B.; Schaefer, D.C. Stability of individual differences in cardiovascular reactivity. Physiol. Behav. 1978, 21, 675–678. [Google Scholar] [CrossRef]
- Carnevali, L.; Nalivaiko, E.; Sgoifo, A. Respiratory patterns reflect different levels of aggressiveness and emotionality in Wild-type Groningen rats. Respir. Physiol. Neurobiol. 2014, 204, 28–35. [Google Scholar] [CrossRef]
- Falkner, A.L.; Wei, D.; Song, A.; Watsek, L.W.; Chen, I.; Chen, P.; Feng, J.; Lin, D. Hierarchical Representations of Aggression in a Hypothalamic-Midbrain Circuit. Neuron 2020, 106, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Zhou, C.; Shi, S.; Fang, L.; Liu, J.; Sun, D.; Jiang, L.; Zhang, S. Weighted single-step genome-wide association study of semen traits in Holstein bulls of China. Front. Genet. 2019, 10, 1053. [Google Scholar] [CrossRef] [Green Version]
- Imumorin, I.G.; Kim, E.H.; Lee, Y.M.; De Koning, D.J.; van Arendonk, J.A.; Donato, M.D.; Taylor, J.F.; Kim, J.J. Genome scan for parent-of-origin QTL effects on bovine growth and carcass traits. Front. Genet. 2011, 2, 44. [Google Scholar] [CrossRef] [Green Version]
- Bandler, R.; Keay, K.A. Columnar organization in the midbrain periaqueductal gray and the integration of emotional expression. Prog. Brain Res. 1996, 107, 285–300. [Google Scholar] [PubMed]
- Fineberg, S.K.; Ross, D.A. Oxytocin and the Social Brain. Biol. Psychiatry 2017, 81, e19–e21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, N.J.; Reverter, A.; Greenwood, P.L.; Guo, B.; Café, L.M.; Dalrymple, B.P. Longitudinal muscle gene expression patterns associated with differential intramuscular fat in cattle. Animal 2015, 9, 650–659. [Google Scholar] [CrossRef] [Green Version]
- Roudbari, Z.; Coort, S.L.; Kutmon, M.; Eijssen, L.; Melius, J.; Sadkowski, T.; Evelo, C.T. Identification of biological pathways contributing to marbling in skeletal muscle to improve beef cattle breeding. Front. Genet. 2020, 10, 1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sewalem, A.; Miglior, F.; Kistemaker, G.J. Short communication: Genetic parameters of milking temperament and milking speed in Canadian Holsteins. J. Dairy Sci. 2011, 94, 512–516. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Li, J.; Bonham, A.J.; Wang, Y.; Deng, H. Gains in power for exhaustive analyses of haplotypes using variable-sized sliding window strategy: A comparison of association-mapping strategies. Eur. J. Hum. Genet. 2009, 17, 785–792. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.G.; Pfeiffer, R.; Gail, M.H. Haplotype analysis in population genetics and association studies. Pharmacogenomics 2003, 4, 171–178. [Google Scholar] [CrossRef]
- Hayes, B.J.; Chamberlain, A.J.; McPartlan, H.; Macleod, I.; Sethuraman, L.; Goddard, M.E. Accuracy of marker-assisted selection with single markers and marker haplotypes in cattle. Genet. Res. 2007, 89, 215–220. [Google Scholar] [CrossRef]
- Calus, M.P.; Meuwissen, T.H.; Windig, J.J.; Knol, E.F.; Schrooten, C.; Vereijken, A.L.; Veerkamp, R.F. Effects of the number of markers per haplotype and clustering of haplotypes on the accuracy of QTL mapping and prediction of genomic breeding values. Genet. Sel. Evol. 2009, 41, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, Z.; Wolc, A.; Su, H.; Fernando, R.L.; Dekkers, J.C.M.; Arango, J.; Settar, P.; Fulton, J.E.; O’Sullivan, N.P.; Garrick, D.J. Identification of recombination hotspots and quantitative trait loci for recombination rate in layer chickens. J. Anim. Sci. Biotechnol. 2019, 10, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, Z.-Q.; Saatchi, M.; Schnabel, R.D.; Taylor, J.F.; Garrick, D.J. Recombination locations and rates in beef cattle assessed from parent-offspring pairs. Genet. Sel. Evol. 2014, 46, 34. [Google Scholar] [CrossRef] [Green Version]
- Khatkar, M.S.; Zenger, K.R.; Hobbs, M.; Hawken, R.J.; Cavanagh, J.A.L.; Barris, W.; McClintock, A.E.; McClintock, S.; Thomson, P.T.; Tier, B.; et al. A Primary Assembly of a Bovine Haplotype Block Map Based on a 15,036-Single-Nucleotide Polymorphism Panel Genotyped in Holstein–Friesian Cattle. Genetics 2007, 176, 763–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Marker Information 1 | Scenario Abbreviation |
|---|---|---|
| ssGWAS | H0.15 | ssGWAS_H0.15 |
| H0.50 | ssGWAS_H0.50 | |
| H0.80 | ssGWAS_H0.80 | |
| NCSNP_H0.15 | ssGWAS_NCSNP_H0.15 | |
| NCSNP_H0.50 | ssGWAS_NCSNP_H0.50 | |
| NCSNP_H0.80 | ssGWAS_NCSNP_H0.80 | |
| WssGWAS iteration 2 (WssGWAS_2) | H0.15 | WssGWAS_2_H0.15 |
| H0.50 | WssGWAS_2_H0.50 | |
| H0.80 | WssGWAS_2_H0.80 | |
| NCSNP_H0.15 | WssGWAS_2_NCSNP_H0.15 | |
| NCSNP_H0.50 | WssGWAS_2_NCSNP_H0.50 | |
| NCSNP_H0.80 | WssGWAS_2_NCSNP_H0.80 | |
| WssGWAS iteration 3 (WssGWAS_3) | H0.15 | WssGWAS_3_H0.15 |
| H0.50 | WssGWAS_3_H0.50 | |
| H0.80 | WssGWAS_3_H0.80 | |
| NCSNP_H0.15 | WssGWAS_3_NCSNP_H0.15 | |
| NCSNP_H0.50 | WssGWAS_3_NCSNP_H0.50 | |
| NCSNP_H0.80 | WssGWAS_3_NCSNP_H0.80 |
| Descriptive | LD_0.15 | LD_0.50 | LD_0.80 |
|---|---|---|---|
| Number of non-clustered SNPs | 20,849 | 30,501 | 36,811 |
| Number of clustered SNPs | 21,784 | 12,132 | 5822 |
| Number of blocks | 9634 | 5617 | 2721 |
| Minimum number of SNP in blocks | 2 | 2 | 2 |
| Maximum number of SNP in blocks | 9 | 7 | 7 |
| Average (SD 1) block size (Mb) | 0.035 (0.020) | 0.032 (0.014) | 0.030 (0.013) |
| Minimum block size (bp) | 65 | 65 | 84 |
| Maximum block size (Mb) | 0.201 | 0.161 | 0.160 |
| Number of pseudo-SNPs 2 before QC | 56,734 | 27,324 | 12,877 |
| Number of pseudo-SNPs after QC | 44,559 | 23,918 | 11,389 |
| Number of non-clustered and pseudo-SNPs before QC | 77,583 | 57,825 | 49,688 |
| Number of non-clustered and pseudo-SNPs after QC | 65,435 | 54,444 | 48,227 |
| Chromosome | Gene | S_P (Mb) 2 | E_P (Mb) 3 | GO_BP | KEGG |
|---|---|---|---|---|---|
| BTA5 | ATXN10 | 116.029 | 116.169 | GO:0031175 | - |
| BTA10 | ADAM10 | 51.536 | 51.679 | - | bta05010 |
| BTA11 | VAX2 | 13.483 | 13.509 | GO:0007409, GO:0007601, GO:0030900, GO:0048048, GO:0060041 | - |
| BTA11 | ATP6V1B1 | 13.454 | 13.480 | GO:00076605, GO:0042472 | - |
| BTA14 | CRISPLD1 | 38.295 | 38.346 | GO:0060325 | - |
| BTA15 | CAPRIN1 | 64.662 | 64.697 | GO:0050775, GO0061003 | |
| BTA18 | FA2H | 2.151 | 2.206 | GO:0032286, GO:0032287 | - |
| BTA20 | SPEF2 | 38.369 | 38.573 | GO:0048702, GO:0048854, GO:0069541 | - |
| BTA22 | PLXNA1 | 60.240 | 60.280 | GO:0021785, GO: 0048841, GO:1902287, GO:1990138 | bta04360 |
| BTA22 | CACNA2D3 | 45.925 | 46.819 | - | bta04921 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Araujo, A.C.; Carneiro, P.L.S.; Alvarenga, A.B.; Oliveira, H.R.; Miller, S.P.; Retallick, K.; Brito, L.F. Haplotype-Based Single-Step GWAS for Yearling Temperament in American Angus Cattle. Genes 2022, 13, 17. https://doi.org/10.3390/genes13010017
Araujo AC, Carneiro PLS, Alvarenga AB, Oliveira HR, Miller SP, Retallick K, Brito LF. Haplotype-Based Single-Step GWAS for Yearling Temperament in American Angus Cattle. Genes. 2022; 13(1):17. https://doi.org/10.3390/genes13010017
Chicago/Turabian StyleAraujo, Andre C., Paulo L. S. Carneiro, Amanda B. Alvarenga, Hinayah R. Oliveira, Stephen P. Miller, Kelli Retallick, and Luiz F. Brito. 2022. "Haplotype-Based Single-Step GWAS for Yearling Temperament in American Angus Cattle" Genes 13, no. 1: 17. https://doi.org/10.3390/genes13010017