Genomics and Therapeutic Vulnerabilities of Primary Bone Tumors
by
 , , and
, , and
Katia Scotlandi
1,* ,
,
Claudia Maria Hattinger
1, 
Evelin Pellegrini
1,
Marco Gambarotti
2 and
Massimo Serra
1,* 1
Laboratory of Experimental Oncology, IRCCS Istituto Ortopedico Rizzoli, via di Barbiano 1/10, 40136 Bologna, Italy
2
Department of Pathology, IRCCS Istituto Ortopedico Rizzoli, via di Barbiano 1/10, 40136 Bologna, Italy
*
Authors to whom correspondence should be addressed.
Cells 2020, 9(4), 968; https://doi.org/10.3390/cells9040968
Submission received: 19 March 2020
/
Revised: 8 April 2020
/
Accepted: 10 April 2020
/
Published: 14 April 2020
(This article belongs to the Special Issue Molecular and Cellular Mechanisms of Cancers: Bone Sarcomas)
Abstract
:Osteosarcoma, Ewing sarcoma and chondrosarcoma are rare diseases but the most common primary tumors of bone. The genes directly involved in the sarcomagenesis, tumor progression and treatment responsiveness are not completely defined for these tumors, and the powerful discovery of genetic analysis is highly warranted in the view of improving the therapy and cure of patients. The review summarizes recent advances concerning the molecular and genetic background of these three neoplasms and, of their most common variants, highlights the putative therapeutic targets and the clinical trials that are presently active, and notes the fundamental issues that remain unanswered. In the era of personalized medicine, the rarity of sarcomas may not be the major obstacle, provided that each patient is studied extensively according to a road map that combines emerging genomic and functional approaches toward the selection of novel therapeutic strategies.
1. Introduction
Bone sarcomas are rare diseases, with an overall annual incidence of 1 case per 100,000 adults in Europe, and they represent 2% of all human neoplasms. They include diverse mesenchymal malignancies that arise from bone, cartilage and connective tissues, representing a very heterogenous group of tumors which range from indolent to very aggressive and metastatic. The World Health Organization (WHO) has recently reviewed the classification of sarcomas based on both the established histological features and molecular alterations [1]. From a molecular/genetics point of view, sarcomas have traditionally been classified into two major categories, the first including sarcomas with simple, near-diploid karyotypes and simple, translocation-associated alterations, and the second comprising tumors with complex and unbalanced karyotypes, characterized by multiple genomic aberrations. A detailed listing of the genetic features in sarcomas included in the two categories is available in the excellent previous reviews [2,3]. These categories, however, do not fully reflect the genetic diversity of the different tumors. The disposal of widespread genome- and epigenome-wide profiling has just started to reveal how heterogeneous sarcomas are at the molecular/genetic level and has resulted in the identification of reliable diagnostic and prognostic/predictive factors and novel guidance for medical treatments. So far, treatment of sarcomas has historically been rather similar for all the subtypes, comprising conventional chemotherapy, surgery and irradiation, and it is hoped that the application of this rapidly expanding knowledge not only refines diagnosis but also impacts the way in which clinical studies are designed and conducted. This review summarizes the current state-of-art molecular comprehension of primary bone tumors, and how this information is being translated into novel, more personalized, risk-based clinical studies. Particularly, the review highlights the molecular mechanisms of sarcomagenesis for the three most common malignant primary tumors of bone: osteosarcoma, Ewing sarcoma and chondrosarcoma, as paradigms of sarcomas typified by complex molecular alterations and genome instability, of translocation-associated sarcomas and of sarcomas whose natural histories’ include stages of tumor progression, respectively. The review also emphasizes the advances in genomics for a better understanding of sarcomagenesis, diagnosis and therapy of these tumors and notes the fundamental issues that remain unanswered.
2. Osteosarcoma
Osteosarcomas are malignant tumors composed of mesenchymal cells producing osteoid and immature bone. Based on their anatomo-clinical presentation, treatment and prognosis, high-grade varieties (90%) can be distinguished from low- and intermediate-grade varieties of osteosarcomas (10%) [4]. Most genetic studies have been performed on the so-called conventional high-grade osteosarcoma (HGOS), which is localized in the extremities, nonmetastatic at the clinical onset and arising in patients younger than 40 years.
Genetic characterization of HGOS has evolved during the last decade thanks to the integration of conventional and new generation candidate-driven and genome-wide technologies. The highly heterogeneous genetic background of HGOS opposes the identification of molecular and genetic biomarkers. However, several studies have highlighted candidate genetic markers, which can be translated into clinics in the near future (Figure 1), whereas other candidate targets have been recently considered to launch several clinical trials (Table 1).
2.1. Low-Grade Osteosarcomas
Low-grade osteosarcomas (parosteal and low-grade central osteosarcoma) and intermediate-grade osteosarcomas (periosteal) are very rare and grow slower than high-grade varieties of osteosarcomas. Removal of the tumor by surgery with wide margins is the gold standard of therapy. Prognosis is good unless a progression to a high-grade osteosarcoma occurs. Chemotherapy with the same drugs as used for HGOS is indicated only in patients with dedifferentiated parosteal osteosarcoma or low-grade central osteosarcoma progressed in HGOS [4].
Different to HGOS, on which the following sections will focus, low-grade osteosarcoma varieties (parosteal and low-grade central) frequently harbor amplifications of the 12q13-15 amplicon, including the MDM2 and CDK4 genes, as supernumerary ring or giant chromosomes. In a recent study of 22 low-grade osteosarcomas (3 low-grade central, 14 classic parosteal, and 5 dedifferentiated parosteal osteosarcomas), an MDM2 (12q15) amplification was revealed in 100% of the cases by fluorescence in situ hybridization. In addition, the fibroblast growth factor receptor substrate 2 (FRS2) gene at 12q15 was amplified in 95% of the cases but not in their histological mimics [5].
2.2. High-Grade Osteosarcomas
2.2.1. Germline Genetics
Germline genetics have tried to identify common variants that may be associated with HGOS and to identify the genetic origin of osteosarcoma [6]. HGOS is associated with several rare and autosomal cancer predisposition syndromes, including the Li–Fraumeni syndrome (mutations of the TP53 gene), hereditary retinoblastoma (mutations of the RB1 gene), Bloom syndrome (mutations of the RECQL3 gene), Werner syndrome (mutations of the RECQL2 (WRN) gene), Rothmund–Thomson, Baller–Gerold and RAPADILINO syndromes (mutations of the RECQL4 gene). Candidate gene studies and, more recently, studies using high-throughput technologies like genome-wide association or next-generation sequencing (NGS) have been performed and have identified promising genes and pathways possibly involved in the aetiology of HGOS [6]. Additional rare variants were found in five cancer predisposition genes, APC, MSH2, PALB2, SQSTM1 and ERCC2, and in common variants in more than 20 genes belonging to bone homeostasis, growth, DNA repair-related, apoptosis, cell cycles, detox reactive oxidative species and tumor immunity pathways [6].
2.2.2. Somatic Genetics
HGOS is characterized by a hyperdiploid, frequently a near-tri- to -tetraploid, an extremely complex karyotype displaying many copy number and structural aberrations with the result of losses and gains of the chromosomal fragments but without any pathognomonic chromosomal alteration [7]. The high genomic instability can be ascribed to chromothripsis (a massive genomic rearrangement due to a cataclysmic event in which chromosomes are fragmented and subsequently aberrantly assembled) and kataegis (high number of genetic changes due to localized hypermutation areas), both frequently described in HGOS [6,8]. A study focusing on somatic copy number alterations in 160 HGOS cases even indicated chromothripsis and near-tetraploidy associated with worse survival [9].
The results obtained in the studies using genome-wide NGS techniques have been discussed in two complete reviews [6,8]. Facing the genetic complexity and heterogeneity of HGOS, concomitantly with its rarity, is challenging. However, Rickel et al. provided a list of 53 candidate onco- and tumor-suppressor genes with validated somatic mutations identified in three studies [10,11,12]. All these genes belong to cancer signaling pathways regulating cell fate and survival and genome maintenance.
In order to better understand the genetic mechanisms of progression and evolution in HGOS, matched primary and normal samples from 13 pediatric patients with HGOS and matched metastatic or local recurrence samples of the same patients were analyzed by whole-genome sequencing and integrative analysis [13]. The data confirmed a highly heterogeneous mutational landscape including the presence of hypermutation and microsatellite instability. Recurrent somatic alterations were not found, except for structural variations (SVs) of TP53 in 9 out of 13 patients. Multiple germline alterations were found in 21 genes known to be implicated in cancer in 12 cases, but not of TP53. Copy number alterations showed higher conservation in metastases, whereas short variants were less conserved. The kinase insert domain receptor (KDR) gene was found to be commonly amplified and overexpressed in advanced cases and associated with a poor prognosis.
2.2.3. Candidate Predictive and Prognostic Biomarkers
Single nucleotide polymorphisms (SNPs) in several candidate genes of possible relevance for the biology, treatment response and drugs activation or detoxification of HGOS have been evaluated in order to indicate markers predictive of tumor progression, therapy response or patients’ susceptibility to develop treatment-related toxicities [14,15].
The candidate genes that have been most frequently studied in HGOS belong to DNA repair and methotrexate metabolism. However, it must be underlined that most studies were performed on a small patient series, and therefore the reported findings need to be further validated in a prospective way. Concerning DNA repair, the most consistent data have been reported for polymorphisms of the excision repair 1 (ERCC1) and excision repair 2 (ERCC2) genes, both involved in the nucleotide excision repair (NER) pathway. Although findings have sometimes been contradictory, two meta-analyses indicated that HGOS patients carrying the C allele of ERCC1 rs11615 [16,17] or the A allele of ERCC2 rs1799793 [17] have higher probabilities of responding to conventional chemotherapy among Chinese populations. Among genes related to methotrexate metabolism, the most widely studied in HGOS has been the 5,10-methylenetetrahydrofolate reductase (MTHFR) [15,18]. The most remarkable information on the possible clinical impact about this gene consisted with the association of the variant allele (T) of MTHFR rs1801133 with higher degrees of liver and hematologic toxicities in a series of 59 Han Chinese HGOS patients [19].
NGS approaches are also expected to provide insights leading to the identification of the HGOS driver genes, which may not consistently emerge with conventional techniques. The application of NGS and other genome-wide analyses to HGOS has further confirmed the presence of multiple genomic rearrangements, kataegis and a high degree of intratumor heterogeneity [10,20], but it has also indicated somatic gene mutations that appear to drive HGOS oncogenesis. One of the most clinically relevant information derived from the NGS and genome-wide studies applied to HGOS is the indication that the phosphatidylinositol 3-kinase/mammalian target of the rapamycin (PI3K/mTOR) pathway can be considered as a key target for HGOS treatment, providing the rationale for designing mTOR-targeting clinical trials [12,21,22] (Table 1). Phase I/II trials have been thus launched for patients with advanced soft-tissue and bone sarcomas (including HGOS) to assess the clinical efficacy of different mTOR inhibitors: temsirolimus (CCI-779, Torisel®, Wyeth Pharmaceuticals, Midrand, South Africa), everolimus (RAD-001; Novartis, Basel, Switzerland), deferolimus (AP23573, Ariad Pharmaceutical, Cambridge, MA, USA) and ridaforolimus (AP23573/MK-8669, Merck, Kenilworth, NJ). Although these drugs have shown encouraging antitumor activities and good tolerability, their therapeutic value for HGOS treatment still needs to be defined [12,21]. However, in a phase II trial performed by the Italian Sarcoma Group (ISG), the activity of everolimus in combination with sorafenib was evaluated in patients with inoperable HGOS, who progressed after conventional chemotherapy treatments. Despite that this study did not achieve the planned target of a 6-month progression-free survival in at least 50% of patients, 17 of the 38 enrolled patients did not present any tumor progression at 6 months, encouraging the use of this drug combination as a rescue treatment for patients with advanced or unresectable HGOS [23].
Another NGS finding with significant clinical relevance in HGOS, is the indication that this tumor frequently exhibits deficiencies in the BRCA1- and/or BRCA2-pathway (the so-called BRCAness phenotype). This body of information may constitute the basis for a future tailoring of HGOS treatment, since the BRCA1/2 pathway plays a key role in the repair of DNA damages via the homologous recombination pathway, an impairment associated with the BRCAness phenotype that may render these HGOS patients more sensitive to targeted therapy with poly(ADP-ribose) polymerase (PARP) inhibitors [11].
In the last few years, coordinated programs and multi-institutional pipelines have been developed to translate the NGS-generated body of information into clinical applications for several tumors, including HGOS. For example, the individualized therapy for relapsed malignancies in childhood (INFORM) program (http://pediatric-neurooncology.dkfz.de/index.php/en/diagnostics/inform) is a multicentric study, the objective of which is the identification of therapeutic targets for children with malignant tumors, including HGOS [24]. The therapeutically applicable research to generate effective treatments (TARGET) project (ClinicalTrials.gov identifier: NCT01190943; http://ocg.cancer.gov/programs/target/projects/osteosarcoma) aims for the identification of genetic and epigenetic alterations of possible clinical relevance in HGOS through a combination of different genomic approaches. Recently, another strategy, consisting of the integration of genomic copy number variations and chemotherapy response-related biomarkers, has provided insights that may be of help to tailor therapy in pediatric patients with bone and soft-tissue sarcomas on the basis of individual genomic data [25].
2.2.4. Epigenetics
In addition to changes in DNA sequence, heritable epigenetic alterations targeting the whole genome can also modulate the expression of many genes, including those involved in important biologic processes such as genomic stability, DNA repair, cell cycle control and apoptosis, among others [26].
Like for other human tumors, several evidence have reported the impact of epigenetic regulation on gene expression also in HGOS [27,28]. In particular, promoter hypermethylation was indicated as the main cause for the decreased expression of 384 genes which are physiologically down-regulated in human embryonic stem cells, suggesting that this mechanism may be a means through which HGOS cells dedifferentiate and become similar to the pluripotent stem cells of their normal tissue of origin [29]. A quantitative assessment of promoter hypermethylation in multiple genes was performed in 30 paired samples of HGOS and normal tissues, revealing a significantly higher incidence of hypermethylation for several gene promoters in tumors compared to normal tissues [30]. The genes that showed the most consistent differences in promoter hypermethylation were the TIMP metallopeptidase inhibitor 3 (TIMP3), death-associated protein kinase 1 (DAPK1) and O-6-methylguanine DNA methyltransferase (MGMT). The downregulation of TIMP3 and DAPK1 genes may impact the resistance to apoptosis or tumor cell invasion [31,32,33], whereas that of MGMT may negatively affect the efficiency of DNA repair [34]. Other evidence indicated that the promoter hypermethylation of the Ras association domain family 1A (RASSF1A) gene, a tumor-suppressor gene involved in cell cycle arrest, microtubule stabilization and apoptosis, is found to be higher in HGOS, compared with normal tissues [30]. The expression of the APC down-regulated 1 (APCDD1) gene, an inhibitor of the Wnt pathway that is frequently activated in HGOS, was also demonstrated to be down-regulated through hypermethylation of its promoter in HGOS, with a consequent enhancement of tumor invasion and metastatic properties [35]. Finally, promoter hypermethylation of the cyclin-dependent kinase Inhibitor 2A (CDKN2A) gene, which acts as tumor-suppressor by regulating cell cycles through encoding the p16INK4a and p14ARF proteins, has also been reported in HGOS and suggested to be correlated with metastasis development [30].
Despite the loss of function of the TP53 and RB1 tumor-suppressor genes in HGOS that are mostly a consequence of genetic alterations, it has been reported that the expression of these genes can be dysregulated also through epigenetic mechanisms [28]. Moreover, recent findings have revealed promoter hypermethylation of the genes belonging to the TP53 pathway in HGOS experimental models and clinical samples, indicating that this epigenetic mechanism may induce the impairment of TP53-related activities [28]. In another study, down-regulation of the p14ARF protein (which inhibits MDM2, thus influencing TP53 function), as a consequence of the p14ARF gene promoter hypermethylation, was detected in 15 out of 32 HGOS clinical samples and proved to be associated with a poor survival [36]. Some authors suggested that the impact of TP53 mutations in the development of at least a subset of HGOS may be complemented by the reduced expression and loss of function of the HIC ZBTB transcriptional repressor 1 (HIC1) gene, consequent to the hypermethylation of its promoter, which was detected in the different series of the HGOS clinical samples [37,38].
The hypermethylation of the RB1 gene promoter has also been detected in HGOS, but its relevance for RB1’s loss of function remains to be established [27,39,40]. On the other hand, promoter hypermethylation of genes belonging to the RB1 pathway has been suggested to be involved in the decrease in RB1-related functions [27,40] Indeed, two studies revealed hypermethylation of the p16INK4a gene promoter, which was associated with a negative or decreased p16INK4a expression, and may impair the RB1-mediated control of cell cycles and tumor cell growth in a minority of HGOS [41,42,43,44].
Gene expression is also regulated by different RNA subclasses of non-coding RNAs, which include small non-coding RNAs (maximum length of 200 base pairs), long non-coding RNAs (lncRNAs, with length greater than 200 base pairs) and circular RNAs (small closed circular RNAs). The majority of data collected so far in HGOS concerns microRNAs (miRNAs) and lncRNAs. For details please refer to recent reviews [45,46,47]. Although the expression patterns and clinical impact of miRNAs and lncRNAs in HGOS largely need to be confirmed and better defined, on the basis of the findings reported so far, it is possible to consider them as novel attractive candidate biomarkers for monitoring disease progression, predicting outcomes and as novel candidate therapeutic targets which might be taken into consideration for planning innovative strategies of therapeutic intervention. Indeed, epigenetic regulators (first of all non-coding RNAs) were found to be involved in the reversion of tumor cells’ malignancy and the induction of cell differentiation [48]. Differentiation-based therapies would be highly desirable for tumors resistant to traditional treatments and could be a promising option for HGOS therapy, considering that the induction of differentiation in HGOS is expected to revert malignancy toward a low-grade osteosarcoma phenotype, but also to reduce tumor cells’ proliferation activity. Recently, it has been demonstrated that the inhibition of DNA methyltransferases (DNMTs), which regulate the process of DNA methylation [49], by novel nonnucleoside inhibitors, affected HGOS tumor proliferation and induced osteoblastic differentiation through the specific re-expression of genes regulating this physiologic process [50,51]. Although differentiated cells, being not or less proliferating, are thought to be less sensitive to cytotoxic drugs, Manara et al. [50] demonstrated that the novel nonnucleoside DNMT inhibitor MC3343 increased the stable doxorubicin bonds to DNA, and combined treatment resulted in sustained DNA damage and increased cell death, thus indicating a potential therapeutic option for patients with HGOS who respond poorly to neoadjuvant chemotherapy.
2.2.5. Immunotherapy and Tumor Mutational Burden
Genomic research in HGOS may also provide relevant information for immunotherapeutic approaches. Immunotherapy has generated many expectancies for increasing cure probability in several cancers but, unfortunately, preclinical and clinical studies using immune checkpoint blockades led to limited and unsatisfactory results in HGOS. Several investigations have been performed or are still ongoing in order to find possible explanations for the reduced efficacy of immunotherapy in HGOS (for a review see [52]). NGS analysis of resected tumor tissues revealed an amplification of PD-L1 and PD-L2, which is associated in the literature with responses to anti-PD-1/PD-L1 blockades in other tumor types and in osteosarcoma mouse models [53]. However, data reporting on the level of the PD-1/PD-L1 expression on neoplastic HGOS cells in paraffin-embedded samples are extremely variable [54,55,56,57]. In general, the PD-L1 expression in HGOS was reported to be present only in a minority of tumor cells, partly explaining the reduced efficacy of immuno checkpoint blockade ICB in HGOS.
In addition, bone sarcomas generally exhibit a low tumor mutational burden (TMB), a predictive biomarker of anti-PD-1/PD-L1 immunotherapy responses [58,59]. Although genetic aberrations are frequent in HGOS, its TMB can be defined as low to moderate, with an average of only seven (or little more) neoepitopes per tumor [60,61], a number that, very likely, is not sufficient to efficiently stimulate the immune system and to determine a good response to anti-PD-1/PD-L1 treatments. Successful immunotherapy based on PD-1/PD-L1 blockades has so far been associated with a high TMB, which facilitates the expression of neoantigens initiating antigen-specific immune responses following immune checkpoint blockade treatment [52,58,62], and with the expression of immune checkpoint molecules. The absence of these conditions in HGOS may well explain the poor effectiveness of ICB in HGOS, supporting the need for more intense studies for a clear identification of the immune infiltrate of these tumors before scheduling the introduction of immunotherapy.
3. Chondrosarcoma
Chondrosarcomas are malignant mesenchymal tumors of bone which produce a cartilaginous matrix. They are the second most common primary malignant bone tumors and usually affect adults with peak incidences in the fifth to seventh decades of life. This category comprises different entities, ranging from locally-aggressive to high-grade malignant neoplasms (for a review see [63,64]. The vast majority (around 90%) are central and secondary peripheral chondrosarcomas, either presenting as de novo in the medulla of bone, or arising as a secondary tumor from pre-existing benign lesions such as enchondromas in enchondromatosis and osteochondromas. The remaining 10% are rare variants of chondrosarcomas, which include periosteal, clear cell, mesenchymal and synovial chondrosarcomas.
Although not mandatory for the diagnosis, genetic alterations characteristic of specific histotypes can be useful in the differential diagnosis with other neoplasms, in particular chondroblastic osteosarcoma. Moreover, the functional study of these alterations can help in understanding the pathogenesis of chondrosarcomas and in the design of a new specific therapy, an urgent unmet medical need, considering that chondrosarcomas are generally not responsive to standard chemotherapy or radiation therapy [65].
Genetically, two main groups of chondrosarcomas exist: central chondrosarcomas, characterized by mutations in the isocitrate dehydrogenase genes IDH1 and IDH2, and secondary peripheral chondrosarcomas, characterized by alterations in the exostosin glycosyltransferase 1 (EXT1) and 2 (EXT2) genes.
3.1. Central Chondrosarcomas
The central atypical cartilaginous tumor/chondrosarcoma grade 1 (ACT/CS1) is a locally aggressive hyaline cartilage-producing tumor arising in the medullary bone. Genetically, primary ACT/CS1 is characterized by somatic mutations in IDH1 and IDH2. These mutations are present in about 50% of cases. [66]. Hotspot mutations are present at the IDH1 p.Arg132 and the IDH2 p.Arg172 positions, the former being the most frequent. Mutations at the IDH1 p.Arg140 position have also been reported [67]. Grade 2 and 3 central chondrosarcomas are intermediate- and high-grade malignant tumors, respectively. Similarly, to ACT/CS1, about 50% of cases show IDH1 or IDH2 mutations. The incidence was found to be higher, up to 80%, in patients with Ollier disease and Maffucci syndrome [66], rare bone diseases characterized by multiple enchondromatosis and an increased risk of malignancies, including pancreatic adenocarcinoma, brain glioma and chondrosarcoma [68]. IDH is an enzyme of the Krebs’s cycle which catalyzes isocitrate to α-ketoglutarate. Somatic mutations in the IDH genes (IDH1 and IDH2) were first discovered in human glioblastomas and are associated with better overall survival [69]. Recently, hotspot sequencing of the IDH1 and IDH2 genes in 89 central chondrosarcomas followed by targeted NGS in 54 of them indicated that IDH1/IDH2 mutations are not associated with the overall survival of patients [70]. However, IDH1/IDH2 mutations were found to be associated with longer relapse-free survival and metastasis-free survival in high-grade chondrosarcomas [70].
Grade 2 and 3 central chondrosarcomas, including dedifferentiated chondrosarcomas are also characterized by complex karyotypes with aneuploidy [39,71]. Other genetic alterations described in the literature are alterations of the RB1 pathway (86% of cases) with a loss of p16 and an overexpression and/or amplification of CDK4, a mutation of TP53 (in up to 59% of the cases) [72] and mutations in COL2A1 (in 45% of cases) [73,74], and also mutations in YEATS2 (12%), EGFR (19%), NRAS (12%), and IHH signaling (18%) [67,74,75]. In about 75% of cases, CDKN2A copy number variations are present. These alterations are absent in enchondroma and ACT/CS1 [76].
Dedifferentiated chondrosarcoma is the most aggressive entity of this group. Genetically, it is characterized by mutations in the IDH1 or IDH2 genes in 50–87% of cases. These alterations are evident in both the areas of conventional chondrosarcoma and in the high-grade dedifferentiated component [77]. IDH1 and IDH2 active site mutations result in the loss of the production of α-ketoglutarate (α-KG) and an accumulation of 2-hydroxyglutarate (2-HG) at supraphysiological levels within cells [78]. An elevated 2-HG level competitively inhibits histone lysine demethylases [79] and the TET family of 5-methylcytosine hydroxylase [80], which results in genome-wide histones and DNA methylation alterations (Figure 2). Thus, mutations in IDH1 and IDH2 may change the expression of potentially large numbers of genes, contributing to tumorigenesis through the alteration of the epigenetic control of gene expression in the cell of origin.
Selective IDH inhibitors which suppress 2-HG production and induce antitumor responses in cells with IDH1 and IDH2 mutations were developed and validated in preclinical settings. Inhibitors of mutated IDH1/2 enzymes (Figure 2) entered clinical trials for targeted therapy of gliomas [81] and may represent an interesting opportunity also for patients with chondrosarcomas. Death is rare in low-grade chondrosarcomas, while it occurs in 30% and 60% of central grade II and grade III tumors, respectively [64], encouraging claims for novel treatment strategies. Amplification of the receptor tyrosine kinases such as IGF1R and KIT which might potentially be targetable by tyrosine kinase inhibitors were also found in chondrosarcoma [70]. In addition, other pathways which might be targets for specific treatments are Hedgehog [82], mTOR [83], SRC and AKT [84]. Some targeted clinical trials are presently ongoing in chondrosarcomas (Table 2). Additional potential targets for therapy in mesenchymal, clear cell and dedifferentiated chondrosarcomas include the Bcl-2 family members and TGFβ genes as potential targets [85]. In particular, the Bcl-2 family seems to play a role in the chemoresistance of chondrosarcomas [86].
3.2. Secondary Peripheral Chondrosarcomas
The secondary peripheral atypical cartilaginous tumor/chondrosarcoma grade 1 (ACT/CS1) is a locally aggressive neoplasm arising within the cartilaginous cap of an osteochondroma. While in osteochondroma, EXT1 or EXT2 are biallelically inactivated in at least a subset of the tumor cells [87], the cartilaginous cap of secondary peripheral ACT/CS1, in tumor progression, becomes gradually populated by cells with at least one functional copy of EXT1 or EXT2 [88], so that EXT-mutant alleles and EXT-wildtype alleles coexist [74]. The proportion of EXT1- or EXT2-mutated alleles among all EXT alleles is about 40%. This supports the hypothesis that genetic factors other than EXT1 or EXT2 mutations, such as CDKN2A [89], are involved in tumor progression. Nevertheless, the loss of heterozygosity in the EXT1 and EXT2 genes has been implicated to cause hereditary multiple exostoses, one of the most common inherited musculoskeletal conditions, with an incidence of 1 in 50,000, whose most serious complication is chondrosarcoma transformation. The presence of germline mutations in the EXT1 or EXT2 genes in patients with multiple osteochondromatosis leads to a 5% risk of developing a secondary peripheral ACT/CS1, compared with only 1% for patients with solitary osteochondromas [90]. EXT is involved in heparan sulphate biosynthesis and the resulting heparan sulphate proteoglycans (HSPGs) are important for cell signaling. In osteochondromas in which the EXT is inactivated, the HSPGs seem to accumulate in the cytoplasm of the cell instead of being transported to be expressed at the cell surface [91], hindering the signaling pathways that are normally operative during skeletal growth, such as the Hedgehog, Wnt and TGF-β signaling pathways (for details see the review [63]). In grade 2 and grade 3 secondary peripheral chondrosarcomas, the EXT-wildtype cells seem to predominate [88]. These tumors also show more complex karyotypes and chromosomal instability. Alterations in the p53 and RB1 pathways have been described [89,92].
3.3. Rare Variants of Chondrosarcoma
Periosteal chondrosarcoma is a rare malignant cartilaginous tumor arising from the bone surface. A subset of these tumors shows IDH1 and IDH2 mutations [66]. Deregulation of the RB1 signaling by the loss of the p16 expression and loss of the Wnt signaling have been described [93].
Clear cell chondrosarcoma is a rare low-grade malignant cartilaginous epiphyseal neoplasm. Clonal abnormalities with diploid or near-diploid complements, losses or structural aberrations of chromosome 9 and gains of chromosome 20 [94], CDKN2A alterations [95] and p53 overexpressions in the absence of detectable mutations [96] have been described. Interestingly, 1/15 clear cell chondrosarcomas investigated for the H3.3 mutations harbor the H3-3B (H3F3B) and p.Lys36Met mutations, suggesting a possible pathogenetic link with chondroblastoma [97].
Mesenchymal chondrosarcoma is a rare high-grade malignant tumor composed of well-differentiated hyaline cartilage and primitive undifferentiated round cells. It is characterized by a specific gene fusion between HEY1 and NCOA2 that occurs in nearly all cases [98]. Since it is absent in other morphologically similar lesions, the molecular detection of this fusion gene is important when differential diagnosis with other neoplasms should be performed. This is particularly relevant in the differential diagnosis with other round cell sarcomas when examining small biopsies of mesenchymal chondrosarcomas lacking the cartilaginous component. A single case report described a case with a IRF2BP2-CDX1 t(1;5)(q42;q32) fusion [99]. Downregulation of the RB1 pathway has also been reported [72].
Finally, synovial chondrosarcoma is an extremely rare malignant tumor, considered the malignant counterpart of synovial chondromatosis. Both these entities are characterized by FN1-ACVR2A and ACVR2A-FN1 fusions [100], which are present in at least 50% of synovial chondromatosis. Since they are present in both benign and malignant forms, the detection of these chimera cannot be used to discriminate between them.
Overall, mutations in IDH1/IDH2 or EXT1/EXT2 have been shown to have a role in the pathogenesis of most common central or peripheral chondrosarcomas, respectively. In addition, inactivation of the Rb and/or p53 pathways are present in most of the tumors and are very likely to have a major role in chondrosarcoma development. More sporadic information indicated that some genes and pathways which are involved in normal cartilage development are disrupted in the chondrosarcoma development. However, only a limited number of studies are available on chondrosarcomas, and for the rare chondrosarcoma subtypes, most genetic studies are merely restricted to case reports.
Research focusing on the elucidation of the molecular events that underlie the pathogenesis of this rare bone malignancy and the identification of new molecularly targeted therapies, especially for chemotherapy refractory chondrosarcomas, is highly desirable.
4. Ewing Sarcoma
Ewing sarcoma, the second most common bone sarcoma in children, is the prototype of sarcomas with simple genetic alterations. It arises more frequently in bone than soft tissues, with a small higher incidence in males compared to females. Histologically, it is characterized by a proliferation of uniform round cells.
The tumor is genetically well-characterized. Its main driver alteration is a specific chromosomal translocation that fuses a member of the FET family of proteins (encoded by FUS, EWSR1 and TAF15), which are RNA-binding proteins involved in transcription and splicing, with different members of the ETS (E26-specific) family of transcription factors, which are involved in cell proliferation, cell differentiation, cell cycle control, angiogenesis and apoptosis. EWS-FLI is the most common chimera (85% of cases) [101]. In the 15–20% of Ewing sarcomas that are negative for EWSR1–FLI1, variant fusions between EWSR1 (or rarely FUS gene) and other members of the ETS family occur, most commonly ERG (encoding transcriptional regulator ERG) followed by ETV1, ETV4, FEV and E1AF [102,103,104,105]. The detection of this specific molecular feature has been integrated in the diagnosis since the 1990s [106]. For details, see the review of this issue from Salguero-Aranda et al. (submitted to journal Cells).
Ewing sarcomas, as other translocation-associated sarcomas and many leukemias, tend to arise de novo, without a clear history of progression. Despite that 8–10% of pediatric cancers are due to germline or mosaic mutations in genes causing cancer predisposition syndromes [107], children at risk for Ewing sarcoma are not clearly defined. EWS is rarely observed in the, approximately, 120 cancer predisposition syndromes described to date [108]. Two recent genome-wide association studies suggest that the interactions between germline variation and somatically acquired EWSR1-FLI1 translocations are important etiologic contributors to EWS risk [109].
The value of the fusion chimera EWS-ets in the genesis of Ewing sarcoma has been widely established. The fusion gene is present at initiation, and it is retained throughout the evolution of the tumor. The chimeric protein is known to cause the transcriptional dysregulation of a relevant number of target genes by inducing de novo enhancers at the GGAA microsatellites (Ewing-specific enhancers) and by repressing enhancers that are active in many cell types, including putative mesenchymal cells of origin [109,110,111]. For a comprehensive review, please refer to [112]. In general, the hybrid product, primarily EWS-FLI, leads to a general remodeling of the cell transcriptome that creates a unique epigenetic signature. The chimera orchestrates multiple oncogenic hits directed toward the disruption of the normal developmental processes and causes the transformation of the cell of origin. However, despite being a necessary condition, the hybrid product is not sufficient to generate a fully transformed phenotype and requires secondary alterations, which may include mutations of the STAG2 (Cohesin subunit SA2) and TP53 genes, which are detected at diagnosis in 15–21% and 5–7% of cases, as well as a deletion of CDKN2A, a cyclin-dependent kinase that regulates cell proliferation, in 10–22% of cases, respectively [113]. Moreover, the presence of an intact IGF pathway as well as of CD99, a 32 kD integral membrane glycoprotein that is peculiarly and highly expressed in Ewing sarcoma cells, is also required for an EWS-FLI transformation [114,115].
Since there are no available drugs that can directly target the fusion proteins, researchers have tackled their downstream pathways and regulatory networks to find alternative targeted therapeutic interventions. This, also in consideration of the higher metastatic potential of EWS cells with a partial silencing of EWS-FLI [116], undermines the rationale for the complete therapeutic suppression of fusion oncoprotein transcripts.
Tumorigenesis in translocation-driven tumors is critically mediated via the IGF-1R signaling pathway [117]. Preclinical models of Ewing sarcomas show an autocrine activation of the IGF-1R-mediated signaling pathway [118], which promotes cell viability and drug escape [119,120]. Over the years, monoclonal antibodies and tyrosine kinase inhibitors were designed to specifically target IGF-1R, and several phase I to III clinical trials were conducted. From these studies, we obtained some important indications: (1) anti-IGF-1R drugs have modest toxic effects and (2) anti-IGF-1R drugs show limited effectiveness on tumors. Due to this disappointing evidence in the big-killer types of cancer, the development of anti-IGF-1R agents was largely abandoned. However, in the Ewing sarcoma clinical studies, a complete response (CR) in a few patients, a partial response (PR) in 2–12% of patients and disease stabilization in 16% to 40% patients were highlighted [121]; [122,123,124]. To date, this is one of the best results obtained in Ewing sarcomas with targeted agents. The combination of IGF-1R inhibitors and other targeted agents (for a review see [125]) or non-specific drugs such as Trabectedin have been proposed. However, despite addressing that the IGF system is still of interest in Ewing sarcomas, there are no recruiting clinical trials today. Efforts aimed to establish predictive response markers are essential for providing new fuel to the field. Besides the evaluation of IGF-1, IGF-2 and IGF-1R levels, the detection of the RNA-binding protein IGF-2-BP3 has been recently suggested [126].
Being an essential molecule for the malignancy of Ewing sarcoma, CD99 is another molecule being studied as a potential target for the treatment of this tumor. CD99 can be easily targeted by antibodies, and the availability of a human bivalent antigen-binding antibody directed against CD99 (dAbd C7) [127] that can efficiently deliver a cell death message in Ewing sarcoma cells while sparing normal cells [128] opens new therapeutic perspectives. Anti-CD99 antibodies exert additive/synergistic effects when combined with conventional agents, such as doxorubicin or vincristine [129], and are effective even against chemoresistant tumor cells. For more details, please refer to the recent reviews [130,131]. Despite these potentialities, the antibodies directed against CD99 have not reached the clinics yet. Recently, Çelik et al. described that the small molecule clofarabine was able to bind to the extracellular portion of CD99, inhibiting the biological properties of Ewing sarcoma cells both in vitro and in vivo [132], thus suggesting a targeted use of an already developed drug.
Developmental pathways and epigenetic modifiers are also of specific interest in fusion-driven sarcomas. Ewing sarcoma is characterized by the paucity of other genetic mutations [113,133,134] and due to the functions of their disease-defining oncogenes, epigenetic dysregulation plays an important role in the maintenance of their phenotype. On this basis, several drugs targeting epigenetic modulators have been tested in preclinical conditions against Ewing sarcoma cells. In particular, drugs against the histone deacetylase (HDAC), an epigenetic regulator of histone methylation [135], the enhancer of zeste homolog 2 (EZH2), a polycomb group protein involved in DNA methylation [136], the bromodomain and extra terminal domain (BET) family, whose members are involved in chromatin regulation, and lysine-specific demethylase 1 (LSD1) have provided interesting data that led to their inclusion in clinical trials. Currently, two phase I trials are recruiting Ewing sarcoma patients to test LSD1 inhibitors (Table 3). Interestingly, the LSD1 inhibitor SP2509 was shown to display synergistic effects with the HDAC inhibitors suberoylanilide hydroxamic acid (SAHA) or romidepsin in vivo [137]. Based on these preclinical studies, more investigations focusing on combinations with different epigenetic inhibitors are warranted.
Finally, the inhibition of EWS-FLI1 binding to RNA helicase A (RHA), which is important for its oncogenic function, was shown to have therapeutic relevance [138,139], while functional, genomic and super-enhancer screenings identified a peculiar sensitivity of Ewing sarcomas to the Cyclin D1/CDK4 pathway [140], and to Poly-ADP-ribose polymerase (PARP) inhibitors [141,142]. Ewing sarcoma patients have been recruited in several trials where these agents have been used in monotherapy [143] or in combination [144,145,146,147].
A list of the presently active targeted clinical trials in Ewing sarcomas is provided in Table 3.
In general, many of these targeted therapies are expected to suffer from the development of resistance. Therefore, the identification of the most effective combination treatments and of adequate schedules may led to the design of innovative, more effective treatments. For example, Salvador-Barbero et al. have recently shown that sequential treatment with CDK4/6 inhibitors following DNA-damaging chemotherapy enhances therapeutic benefits in pancreatic cancer models, despite previous reports that concurrent treatment did not [148].
5. Critical Open Issues and Perspectives
Carrying out molecular studies in patients is necessary, at the best by massive analysis technologies (NGS), to find new potentially actionable molecular targets. This is certainly true for tumors that are still orphans for innovative, targeted drugs. In the era of personalized medicine, the rarity of sarcomas is not a major obstacle, provided that each patient is studied extensively.
Due to the lack of specific, druggable genetic alterations in bone sarcomas, researchers are still forced to work at a discovery level. The application of RNA-seq, a technique that likely reveals biologically informative transcriptional pathways, is the first-line indication. However, it remains unknown how deep the sequencing datasets should be performed in the challenging context of identifying transcriptional signatures that are robustly associated with disease outcome and that may indicate novel therapeutic approaches.
Personalized treatments in refractory patients cannot be effective without knowledge of risk and response. A cross-validation of the results is essential to obtain robust information and high-quality relationships, and international collaboration should be largely encouraged. Collaboration with patients should be stimulated to happen throughout the research process and researchers/clinicians should set the context for patients to contribute meaningfully.
The development of experimental models that may well reflect the biological and genetic diversity of the tumors should be encouraged as an essential step to prioritize but also de-prioritize the therapeutic agents that are raised to the attention of clinicians.
Most patients with bone tumors did not have a known cancer predisposition syndrome. However, for chondrosarcoma and osteosarcoma, a link has been observed, suggesting there might be an unknown genetic susceptibility underlying their development. Investigations of the family history and germline genetic association studies might help elucidate the potential genetic susceptibility in the future.
The possibility of genetic studies over time in the same patient, provided by the study of circulating tumors’ DNA/RNA with highly sensitive techniques, enables the study of prognostic and/or predictive biomarkers, the determination of minimal residual diseases, early detection of resistance and the study of tumor evolution. The application of these analyses will also help patients with predisposition syndromes and should be highly encouraged for pediatric patients
A percentage of patients with bone sarcomas also have histories of second malignancies that are unrelated to bone tumors. A close follow-up of the patient is necessary and appropriate. Detecting a cancer early reduces treatments and treatment-associated morbidity.
Immunotherapy approaches in bone sarcomas have led to disappointing results, so far. The application of massive and parallel analyses is required to determine the specific composition of the immune microenvironment for primary bone sarcomas.
6. Concluding Remarks
The identification of cancer-specific molecular alterations and vulnerabilities is an important objective for bone sarcomas. The detection of tumor-specific molecular alterations is already into routine clinicopathological practice as a molecular tool to assist the pathologist in the differential diagnosis of several histotypes of bone sarcomas. However, a deeper systematic molecular analysis of these lesions is highly warranted to help us to better understand the pathogenesis of these tumors, and to hopefully enable the development of novel and more personalized therapeutic strategies. Early-phase clinical trials guided by comprehensive molecular tumor profiling have become feasible thanks to the reduction of costs for NGS studies and the increased robustness and standardization of procedures. Considering the rarity of these tumors, the definition of a clear, internationally approved road map that combines emerging genomic and functional approaches with a selection of novel therapeutic strategies could provide the basis for transforming the care of patients with bone sarcomas.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
The authors’ studies and findings cited in this manuscript were supported by grants from the Foundation AIRC for Cancer Research (projects AIRC IG2019-ID.22805-P.I. Scotlandi Katia and IG2018-ID.21487-P.I. Serra Massimo).
Acknowledgments
Authors would like to thank Cristina Ghinelli for the help in figure editing. The authors also thank the Associazione “Mario Campanacci” for supporting our activities and all the patients and their families that suffer these diseases for their cooperation to research advances.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The abbreviations including in the text are reported alphabetically.
| ACT/CS1 | atypical cartilaginous tumour/chondrosarcoma grade 1 |
| ACVR2A-FN | activin receptor 2A- fibronectin |
| AKT | Serine/Threonine Kinase+ |
| APC | APC regulator of WNT signaling pathway |
| APCDD1 | APC down-regulated 1 |
| Bcl-2 | Apoptosis Regulator |
| BET | the bromodomain and extra terminal domain |
| BRCA1 | breast related cancer antigen 1 |
| BRCA2 | breast related cancer antigen 2 |
| CD99 | CD99 Molecule (XgBloodGroup) |
| CDK4 | cyclin-dependent kinase 4 |
| CDK4/6 | cyclin-dependent kinase 4/6 |
| CDKN2A | cyclin-dependent kinase Inhibitor 2A |
| CR | complete response |
| DAPK1 | death-associated protein kinase 1 |
| DNA/RNA | DeoxyriboNucleic Acid/RiboNucleic Acid |
| DNMTs | DNA methyltransferases |
| EGFR | epidermal growth factor receptor |
| ERCC1 | excision repair cross-complementation group 1 |
| ERCC2 | excision repair cross-complementation group 2 |
| ERG | ETS transcription factor ERG |
| ETS | proto-oncogene 1, transcription factor 1 |
| ETV1 | ETS Variant Transcription Factor 1 |
| ETV4 | ETS Variant Transcription Factor 4 |
| EWS-FLI | Ewing Sarcoma-Friend Leukemia Insertion |
| EWS | Ewing Sarcoma |
| EWSR1 | Ewing sarcoma breakpoint region 1, EWS RNA Binding Protein 1 |
| EWSR1–FLI1 | Ewing sarcoma breakpoint region 1 (EWS RNA Binding Protein 1) - Friend Leukemia Insertion |
| EXT | exostosin glycosyltransferase |
| EZH2 | enhancer of zeste homolog 2 |
| FEV | FEV Transcription Factor, ETS Family Member |
| FUS | FUS RNA Binding Protein |
| GD2 | disialoganglioside 2 |
| HDAC | histone deacetylase |
| HEY1 | Hes Related Family BHLH Transcription Factor With YRPW Motif 1 |
| HGOS | High-grade osteosarcoma |
| HIC1 | HIC ZBTB transcriptional repressor 1 |
| HSPGs | heparan sulphate proteoglycans |
| ICB | immune checkpoint blockade |
| IDH | isocitrate dehydrogenase genes |
| IGF-1 | Insulin-like growth factor 1 |
| IGF-1R | Insulin-like growth factor 1 receptor |
| IGF-2 | Insulin-like growth factor 2 |
| IGF | Insulin-like growth factor |
| IGF-2BP3 | Insulin-like growth factor 2 - mRNA-binding protein 3 |
| IHH | Indian Hedgehog Signaling Molecule |
| INFORM | individualized therapy for relapsed malignancies in childhood |
| IRF2BP2-CDX1 | Interferon Regulatory Factor 2 Binding Protein 2-Caudal Type Homeobox 1 |
| ISG | Italian Sarcoma Group |
| KDR | kinase insert domain receptor |
| KIT | Proto-Oncogene, Receptor Tyrosine Kinase |
| lncRNA | Long non-coding RNA |
| lncRNAs | long non-coding RNAs |
| LSAMP | Limbic System-Associated Membrane Protein |
| LSD1 | Lysine Demethylase 1° |
| MDM2 | MDM2 Proto-Oncogene |
| MGMT | O-6-methylguanine DNA methyltransferase |
| miRNA | microRNA |
| miRNAs | microRNAs |
| MSH2 | mut S homolog 2 |
| MTHFR | 5,10-methylenetetrahydrofolate reductase |
| mTOR | mammalian target of rapamycin |
| NADPH | reduced nicotinamide adenine dinucleotide phosphate |
| NCOA2 | Nuclear Receptor Coactivator 2 |
| NER | Nucleotide excision repair |
| NGS | Next-generation sequencing |
| NRAS | NRAS Proto-Oncogene |
| p16 | also known as p16INK4a, cyclin-dependent kinase inhibitor 2A or CDKN2A, multiple tumor suppressor 1 |
| PALB2 | partner and localizer of BRCA2 |
| PARP | poly (ADP-ribose) polymerase |
| PD-1 | programmedcelldeath 1 |
| PD-L1 | programmed death-ligand 1 |
| PI3K/mTOR | phosphatidylinositol 3-kinase/mammalian target of rapamycin |
| PR | partial response |
| RASSF1A | Ras association domain family 1A |
| RB1 | RB TranscriptionalCorepressor 1 |
| RECQL | RecQ-like helicase |
| RHA | RNA helicase A |
| SAHA | suberoylanilide hydroxamic acid |
| SNP | Single nucleotide polymorphism |
| SQSTM1 | Sequestosome 1 |
| SRC | SRC Proto-Oncogene, Non-Receptor Tyrosine Kinase |
| STAG2 | Stromal Antigen 2 |
| SV | structural variations |
| TAF15 | TATA-Box Binding Protein Associated Factor 15 |
| TARGET | Generate Effective Treatments Osteosarcoma project |
| TET | Tet Methylcytosine Dioxygenase |
| TGFβ | transforming growth factor β |
| TIMP3 | TIMP metallopeptidase inhibitor 3 |
| TMB | tumor mutational burden |
| TP53 | Tumor Protein P53 |
| WWOX | WW Domain ContainingOxidoreductase |
| α-KG | α-ketoglutarate |
| 2-HG | 2-hydroxyglutarate |
References
- Fletcher, C.D.; Bridge, J.; Hogendoorn, P.C.; Mertens, F. WHO Classification of tumours of Soft Tissue and Bone, 4th ed.; WHO Press: Geneva, Switzerland, 2013; Volume 5. [Google Scholar]
- Taylor, B.S.; Barretina, J.; Maki, R.G.; Antonescu, C.R.; Singer, S.; Ladanyi, M. Advances in sarcoma genomics and new therapeutic targets. Nat. Rev. Cancer 2011, 11, 541–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baroy, T.; Chilamakuri, C.S.; Lorenz, S.; Sun, J.; Bruland, O.S.; Myklebost, O.; Meza-Zepeda, L.A. Genome Analysis of Osteosarcoma Progression Samples Identifies FGFR1 Overexpression as a Potential Treatment Target and CHM as a Candidate Tumor Suppressor Gene. PLoS ONE 2016, 11, e0163859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picci, P. Osteosarcomas (OS). In Diagnosis of Musculoskeletal Tumors and Tumor-Like Conditions, 2nd ed.; Picci, P., Manfrini, M., Donati, D.M., Gambarotti, M., Righi, A., Vanel, D., Dei Tos, A.P., Eds.; Springer: Cham, Switzerland, 2020; pp. 185–212. [Google Scholar]
- He, X.; Pang, Z.; Zhang, X.; Lan, T.; Chen, H.; Chen, M.; Yang, H.; Huang, J.; Chen, Y.; Zhang, Z.; et al. Consistent Amplification of FRS2 and MDM2 in Low-grade Osteosarcoma: A Genetic Study of 22 Cases With Clinicopathologic Analysis. Am. J. Surg. Pathol. 2018, 42, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- Gianferante, D.M.; Mirabello, L.; Savage, S.A. Germline and somatic genetics of osteosarcoma—Connecting aetiology, biology and therapy. Nat. Rev. Endocrinol. 2017, 13, 480–491. [Google Scholar] [CrossRef]
- Sandberg, A.A.; Bridge, J.A. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors: Osteosarcoma and related tumors. Cancer Genet. Cytogenet. 2003, 145, 1–30. [Google Scholar] [CrossRef]
- Rickel, K.; Fang, F.; Tao, J. Molecular genetics of osteosarcoma. Bone 2017, 102, 69–79. [Google Scholar] [CrossRef]
- Smida, J.; Xu, H.; Zhang, Y.; Baumhoer, D.; Ribi, S.; Kovac, M.; von Luettichau, I.; Bielack, S.; O’Leary, V.B.; Leib-Mosch, C.; et al. Genome-wide analysis of somatic copy number alterations and chromosomal breakages in osteosarcoma. Int. J. Cancer 2017, 141, 816–828. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Bahrami, A.; Pappo, A.; Easton, J.; Dalton, J.; Hedlund, E.; Ellison, D.; Shurtleff, S.; Wu, G.; Wei, L.; et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 2014, 7, 104–112. [Google Scholar] [CrossRef] [Green Version]
- Kovac, M.; Blattmann, C.; Ribi, S.; Smida, J.; Mueller, N.S.; Engert, F.; Castro-Giner, F.; Weischenfeldt, J.; Kovacova, M.; Krieg, A.; et al. Exome sequencing of osteosarcoma reveals mutation signatures reminiscent of BRCA deficiency. Nat. Commun. 2015, 6, 8940. [Google Scholar] [CrossRef]
- Perry, J.A.; Kiezun, A.; Tonzi, P.; Van Allen, E.M.; Carter, S.L.; Baca, S.C.; Cowley, G.S.; Bhatt, A.S.; Rheinbay, E.; Pedamallu, C.S.; et al. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc. Natl. Acad. Sci. USA 2014, 111, E5564–E5573. [Google Scholar] [CrossRef] [Green Version]
- Negri, G.L.; Grande, B.M.; Delaidelli, A.; El-Naggar, A.; Cochrane, D.; Lau, C.C.; Triche, T.J.; Moore, R.A.; Jones, S.J.; Montpetit, A.; et al. Integrative genomic analysis of matched primary and metastatic pediatric osteosarcoma. J. Pathol. 2019, 249, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Hattinger, C.M.; Serra, M. Role of pharmacogenetics of drug-metabolizing enzymes in treating osteosarcoma. Expert Opin. Drug Metab. Toxicol. 2015, 11, 1449–1463. [Google Scholar] [CrossRef] [PubMed]
- Serra, M.; Hattinger, C.M. The pharmacogenomics of osteosarcoma. Pharmacogenomics J. 2017, 17, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Z.; Deng, C.; Tian, Y.; Ma, X. Meta-analysis showing that ERCC1 polymorphism is predictive of osteosarcoma prognosis. Oncotarget 2017, 8, 62769–62779. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Ge, J.; Hong, H.; Bi, L.; Sun, Z. Genetic polymorphisms in ERCC1 and ERCC2 genes are associated with response to chemotherapy in osteosarcoma patients among Chinese population: A meta-analysis. World J. Surg. Oncol. 2017, 15, 75. [Google Scholar] [CrossRef] [Green Version]
- Hattinger, C.M.; Tavanti, E.; Fanelli, M.; Vella, S.; Picci, P.; Serra, M. Pharmacogenomics of genes involved in antifolate drug response and toxicity in osteosarcoma. Expert Opin. Drug Metab. Toxicol. 2017, 13, 245–257. [Google Scholar] [CrossRef]
- Xie, L.; Guo, W.; Yang, Y.; Ji, T.; Xu, J. More severe toxicity of genetic polymorphisms on MTHFR activity in osteosarcoma patients treated with high-dose methotrexate. Oncotarget 2018, 9, 11465–11476. [Google Scholar] [CrossRef] [Green Version]
- Egas-Bejar, D.; Anderson, P.M.; Agarwal, R.; Corrales-Medina, F.; Devarajan, E.; Huh, W.W.; Brown, R.E.; Subbiah, V. Theranostic Profiling for Actionable Aberrations in Advanced High Risk Osteosarcoma with Aggressive Biology Reveals High Molecular Diversity: The Human Fingerprint Hypothesis. Oncoscience 2014, 1, 167–179. [Google Scholar] [CrossRef]
- Hattinger, C.M.; Patrizio, M.P.; Magagnoli, F.; Luppi, S.; Serra, M. An update on emerging drugs in osteosarcoma: Towards tailored therapies? Expert Opin. Emerg. Drugs 2019, 24, 153–171. [Google Scholar] [CrossRef]
- Zhou, Q.; Deng, Z.; Zhu, Y.; Long, H.; Zhang, S.; Zhao, J. mTOR/p70S6K signal transduction pathway contributes to osteosarcoma progression and patients’ prognosis. Med. Oncol. 2010, 27, 1239–1245. [Google Scholar] [CrossRef]
- Grignani, G.; Palmerini, E.; Ferraresi, V.; D’Ambrosio, L.; Bertulli, R.; Asaftei, S.D.; Tamburini, A.; Pignochino, Y.; Sangiolo, D.; Marchesi, E.; et al. Sorafenib and everolimus for patients with unresectable high-grade osteosarcoma progressing after standard treatment: A non-randomised phase 2 clinical trial. Lancet Oncol. 2015, 16, 98–107. [Google Scholar] [CrossRef]
- Worst, B.C.; van Tilburg, C.M.; Balasubramanian, G.P.; Fiesel, P.; Witt, R.; Freitag, A.; Boudalil, M.; Previti, C.; Wolf, S.; Schmidt, S.; et al. Next-generation personalised medicine for high-risk paediatric cancer patients—The INFORM pilot study. Eur. J. Cancer 2016, 65, 91–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, L.; Pandya, P.H.; Liu, E.; Chandra, P.; Wang, L.; Murray, M.E.; Carter, J.; Ferguson, M.; Saadatzadeh, M.R.; Bijangi-Visheshsaraei, K.; et al. Integration of genomic copy number variations and chemotherapy-response biomarkers in pediatric sarcoma. BMC Med. Genom. 2019, 12, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.A.; Issa, J.P.; Baylin, S. Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 2016, 17, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ye, Z. Epigenetic alterations in osteosarcoma: Promising targets. Mol. Biol. Rep. 2014, 41, 3303–3315. [Google Scholar] [CrossRef]
- Morrow, J.J.; Khanna, C. Osteosarcoma Genetics and Epigenetics: Emerging Biology and Candidate Therapies. Crit. Rev. Oncog. 2015, 20, 173–197. [Google Scholar] [CrossRef] [Green Version]
- Easwaran, H.; Johnstone, S.E.; Van Neste, L.; Ohm, J.; Mosbruger, T.; Wang, Q.; Aryee, M.J.; Joyce, P.; Ahuja, N.; Weisenberger, D.; et al. A DNA hypermethylation module for the stem/progenitor cell signature of cancer. Genome Res. 2012, 22, 837–849. [Google Scholar] [CrossRef] [Green Version]
- Hou, P.; Ji, M.; Yang, B.; Chen, Z.; Qiu, J.; Shi, X.; Lu, Z. Quantitative analysis of promoter hypermethylation in multiple genes in osteosarcoma. Cancer 2006, 106, 1602–1609. [Google Scholar] [CrossRef] [Green Version]
- Bachman, K.E.; Herman, J.G.; Corn, P.G.; Merlo, A.; Costello, J.F.; Cavenee, W.K.; Baylin, S.B.; Graff, J.R. Methylation-associated silencing of the tissue inhibitor of metalloproteinase-3 gene suggest a suppressor role in kidney, brain, and other human cancers. Cancer Res. 1999, 59, 798–802. [Google Scholar]
- Inbal, B.; Cohen, O.; Polak-Charcon, S.; Kopolovic, J.; Vadai, E.; Eisenbach, L.; Kimchi, A. DAP kinase links the control of apoptosis to metastasis. Nature 1997, 390, 180–184. [Google Scholar] [CrossRef]
- Rosas, S.L.; Koch, W.; da Costa Carvalho, M.G.; Wu, L.; Califano, J.; Westra, W.; Jen, J.; Sidransky, D. Promoter hypermethylation patterns of p16, O6-methylguanine-DNA-methyltransferase, and death-associated protein kinase in tumors and saliva of head and neck cancer patients. Cancer Res. 2001, 61, 939–942. [Google Scholar] [PubMed]
- Pegg, A.E. Mammalian O6-alkylguanine-DNA alkyltransferase: Regulation and importance in response to alkylating carcinogenic and therapeutic agents. Cancer Res. 1990, 50, 6119–6129. [Google Scholar] [PubMed]
- Han, W.; Liu, J. Epigenetic silencing of the Wnt antagonist APCDD1 by promoter DNA hyper-methylation contributes to osteosarcoma cell invasion and metastasis. Biochem. Biophys. Res. Commun. 2017, 491, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.H.; Kim, H.S.; Kim, H.H.; Kim, W.H.; Lee, S.H. Aberrant methylation of p14ARF gene correlates with poor survival in osteosarcoma. Clin. Orthop. Relat. Res. 2006, 442, 216–222. [Google Scholar] [CrossRef]
- Rathi, A.; Virmani, A.K.; Harada, K.; Timmons, C.F.; Miyajima, K.; Hay, R.J.; Mastrangelo, D.; Maitra, A.; Tomlinson, G.E.; Gazdar, A.F. Aberrant methylation of the HIC1 promoter is a frequent event in specific pediatric neoplasms. Clin. Cancer Res. 2003, 9, 3674–3678. [Google Scholar]
- Chen, W.; Cooper, T.K.; Zahnow, C.A.; Overholtzer, M.; Zhao, Z.; Ladanyi, M.; Karp, J.E.; Gokgoz, N.; Wunder, J.S.; Andrulis, I.L.; et al. Epigenetic and genetic loss of Hic1 function accentuates the role of p53 in tumorigenesis. Cancer Cell 2004, 6, 387–398. [Google Scholar] [CrossRef] [Green Version]
- Ropke, M.; Boltze, C.; Neumann, H.W.; Roessner, A.; Schneider-Stock, R. Genetic and epigenetic alterations in tumor progression in a dedifferentiated chondrosarcoma. Pathol. Res. Pract. 2003, 199, 437–444. [Google Scholar] [CrossRef]
- Sakai, T.; Toguchida, J.; Ohtani, N.; Yandell, D.W.; Rapaport, J.M.; Dryja, T.P. Allele-specific hypermethylation of the retinoblastoma tumor-suppressor gene. Am. J. Hum. Genet. 1991, 48, 880–888. [Google Scholar]
- Benassi, M.S.; Molendini, L.; Gamberi, G.; Ragazzini, P.; Sollazzo, M.R.; Merli, M.; Asp, J.; Magagnoli, G.; Balladelli, A.; Bertoni, F.; et al. Alteration of pRb/p16/cdk4 regulation in human osteosarcoma. Int. J. Cancer 1999, 84, 489–493. [Google Scholar] [CrossRef]
- Benassi, M.S.; Molendini, L.; Gamberi, G.; Magagnoli, G.; Ragazzini, P.; Gobbi, G.A.; Sangiorgi, L.; Pazzaglia, L.; Asp, J.; Brantsing, C.; et al. Involvement of INK4A gene products in the pathogenesis and development of human osteosarcoma. Cancer 2001, 92, 3062–3067. [Google Scholar] [CrossRef]
- Tsuchiya, T.; Sekine, K.; Hinohara, S.; Namiki, T.; Nobori, T.; Kaneko, Y. Analysis of the p16INK4, p14ARF, p15, TP53, and MDM2 genes and their prognostic implications in osteosarcoma and Ewing sarcoma. Cancer Genet. Cytogenet. 2000, 120, 91–98. [Google Scholar] [CrossRef]
- Patino-Garcia, A.; Pineiro, E.S.; Diez, M.Z.; Iturriagagoitia, L.G.; Klussmann, F.A.; Ariznabarreta, L.S. Genetic and epigenetic alterations of the cell cycle regulators and tumor suppressor genes in pediatric osteosarcomas. J. Pediatr. Hematol. Oncol. 2003, 25, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Palmini, G.; Marini, F.; Brandi, M.L. What Is New in the miRNA World Regarding Osteosarcoma and Chondrosarcoma? Molecules 2017, 22, 417. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Wang, G.; Zheng, Y.; Hua, Y.; Cai, Z. Long non-coding RNAs in osteosarcoma. Oncotarget 2017, 8, 20462–20475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattinger, C.M.; Patrizio, M.P.; Tavanti, E.; Luppi, S.; Magagnoli, F.; Picci, P.; Serra, M. Genetic testing for high-grade osteosarcoma: A guide for future tailored treatments? Expert Rev. Mol. Diagn. 2018, 18, 947–961. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Rao, C.M. Epigenetics in cancer: Fundamentals and Beyond. Pharmacol. Ther. 2017, 173, 118–134. [Google Scholar] [CrossRef]
- Zeng, Y.; Chen, T. DNA Methylation Reprogramming during Mammalian Development. Genes 2019, 10, 257. [Google Scholar] [CrossRef] [Green Version]
- Manara, M.C.; Valente, S.; Cristalli, C.; Nicoletti, G.; Landuzzi, L.; Zwergel, C.; Mazzone, R.; Stazi, G.; Arimondo, P.B.; Pasello, M.; et al. A Quinoline-Based DNA Methyltransferase Inhibitor as a Possible Adjuvant in Osteosarcoma Therapy. Mol. Cancer Ther. 2018, 17, 1881–1892. [Google Scholar] [CrossRef] [Green Version]
- Zwergel, C.; Schnekenburger, M.; Sarno, F.; Battistelli, C.; Manara, M.C.; Stazi, G.; Mazzone, R.; Fioravanti, R.; Gros, C.; Ausseil, F.; et al. Identification of a novel quinoline-based DNA demethylating compound highly potent in cancer cells. Clin. Epigenet. 2019, 11, 68. [Google Scholar] [CrossRef] [Green Version]
- Dyson, K.A.; Stover, B.D.; Grippin, A.; Mendez-Gomez, H.R.; Lagmay, J.; Mitchell, D.A.; Sayour, E.J. Emerging trends in immunotherapy for pediatric sarcomas. J. Hematol. Oncol. 2019, 12, 78. [Google Scholar] [CrossRef]
- Nuytemans, L.; Sys, G.; Creytens, D.; Lapeire, L. NGS-analysis to the rescue: Dual checkpoint inhibition in metastatic osteosarcoma—A case report and review of the literature. Acta Clin. Belg. 2019, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Xiao, Q.; Zhou, B.; Dai, Z.; Kang, Y. Prognostic Significance of Programmed Death Ligand 1 Expression and Tumor-Infiltrating Lymphocytes in Axial Osteosarcoma. World Neurosurg. 2019, 129, e240–e254. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Simon, J.S.; Grosso, J.F.; Martinez, D.; Pawel, B.R.; Santi, M.; Merchant, M.S.; Geoerger, B.; Hezam, I.; Marty, V.; et al. Assessment of programmed death-ligand 1 expression and tumor-associated immune cells in pediatric cancer tissues. Cancer 2017, 123, 3807–3815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, B.; Ren, T.; Huang, Y.; Sun, K.; Wang, S.; Bao, X.; Liu, K.; Guo, W. PD-1 axis expression in musculoskeletal tumors and antitumor effect of nivolumab in osteosarcoma model of humanized mouse. J. Hematol. Oncol. 2018, 11, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmerini, E.; Agostinelli, C.; Picci, P.; Pileri, S.; Marafioti, T.; Lollini, P.L.; Scotlandi, K.; Longhi, A.; Benassi, M.S.; Ferrari, S. Tumoral immune-infiltrate (IF), PD-L1 expression and role of CD8/TIA-1 lymphocytes in localized osteosarcoma patients treated within protocol ISG-OS1. Oncotarget 2017, 8, 111836–111846. [Google Scholar] [CrossRef] [Green Version]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [Green Version]
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016, 17, e542–e551. [Google Scholar] [CrossRef] [Green Version]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- Chang, T.C.; Carter, R.A.; Li, Y.; Wang, H.; Edmonson, M.N.; Chen, X.; Arnold, P.; Geiger, T.L.; Wu, G.; Peng, J.; et al. The neoepitope landscape in pediatric cancers. Genome Med. 2017, 9, 78. [Google Scholar] [CrossRef]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef]
- Gelderblom, H.; Hogendoorn, P.C.; Dijkstra, S.D.; van Rijswijk, C.S.; Krol, A.D.; Taminiau, A.H.; Bovee, J.V. The clinical approach towards chondrosarcoma. Oncologist 2008, 13, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Donati, D.M.; Bianchi, G. Chondrosarcomas (CHS). In Diagnosis of Musculoskeletal Tumors and Tumor-Like Conditions, 2nd ed.; Picci, P., Manfrini, M., Donati, D.M., Gambarotti, M., Righi, A., Vanel, D., Dei Tos, A.P., Eds.; Springer: Cham, Switzerland, 2020; pp. 157–179. [Google Scholar]
- Grimer, R.J.; Gosheger, G.; Taminiau, A.; Biau, D.; Matejovsky, Z.; Kollender, Y.; San-Julian, M.; Gherlinzoni, F.; Ferrari, C. Dedifferentiated chondrosarcoma: Prognostic factors and outcome from a European group. Eur. J. Cancer 2007, 43, 2060–2065. [Google Scholar] [CrossRef] [PubMed]
- Amary, M.F.; Bacsi, K.; Maggiani, F.; Damato, S.; Halai, D.; Berisha, F.; Pollock, R.; O’Donnell, P.; Grigoriadis, A.; Diss, T.; et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J. Pathol. 2011, 224, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Lugowska, I.; Teterycz, P.; Mikula, M.; Kulecka, M.; Kluska, A.; Balabas, A.; Piatkowska, M.; Wagrodzki, M.; Pienkowski, A.; Rutkowski, P.; et al. IDH1/2 Mutations Predict Shorter Survival in Chondrosarcoma. J. Cancer 2018, 9, 998–1005. [Google Scholar] [CrossRef] [Green Version]
- Verdegaal, S.H.; Bovee, J.V.; Pansuriya, T.C.; Grimer, R.J.; Ozger, H.; Jutte, P.C.; San Julian, M.; Biau, D.J.; van der Geest, I.C.; Leithner, A.; et al. Incidence, predictive factors, and prognosis of chondrosarcoma in patients with Ollier disease and Maffucci syndrome: An international multicenter study of 161 patients. Oncologist 2011, 16, 1771–1779. [Google Scholar] [CrossRef] [Green Version]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.G.; Nafa, K.; Agaram, N.; Zehir, A.; Benayed, R.; Sadowska, J.; Borsu, L.; Kelly, C.; Tap, W.D.; Fabbri, N.; et al. Genomic Profiling Identifies Association of IDH1/IDH2 Mutation with Longer Relapse-Free and Metastasis-Free Survival in High-Grade Chondrosarcoma. Clin. Cancer Res. 2020, 26, 419–427. [Google Scholar] [CrossRef] [Green Version]
- Tallini, G.; Dorfman, H.; Brys, P.; Dal Cin, P.; De Wever, I.; Fletcher, C.D.; Jonson, K.; Mandahl, N.; Mertens, F.; Mitelman, F.; et al. Correlation between clinicopathological features and karyotype in 100 cartilaginous and chordoid tumours. A report from the Chromosomes and Morphology (CHAMP) Collaborative Study Group. J. Pathol. 2002, 196, 194–203. [Google Scholar] [CrossRef]
- Meijer, D.; de Jong, D.; Pansuriya, T.C.; van den Akker, B.E.; Picci, P.; Szuhai, K.; Bovee, J.V. Genetic characterization of mesenchymal, clear cell, and dedifferentiated chondrosarcoma. Genes Chromosomes Cancer 2012, 51, 899–909. [Google Scholar] [CrossRef]
- Tarpey, P.S.; Behjati, S.; Cooke, S.L.; Van Loo, P.; Wedge, D.C.; Pillay, N.; Marshall, J.; O’Meara, S.; Davies, H.; Nik-Zainal, S.; et al. Frequent mutation of the major cartilage collagen gene COL2A1 in chondrosarcoma. Nat. Genet. 2013, 45, 923–926. [Google Scholar] [CrossRef]
- Totoki, Y.; Yoshida, A.; Hosoda, F.; Nakamura, H.; Hama, N.; Ogura, K.; Fujiwara, T.; Arai, Y.; Toguchida, J.; Tsuda, H.; et al. Unique mutation portraits and frequent COL2A1 gene alteration in chondrosarcoma. Genome Res. 2014, 24, 1411–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.X.; van Oosterwijk, J.G.; Sicinska, E.; Moss, S.; Remillard, S.P.; van Wezel, T.; Buhnemann, C.; Hassan, A.B.; Demetri, G.D.; Bovee, J.V.; et al. Functional profiling of receptor tyrosine kinases and downstream signaling in human chondrosarcomas identifies pathways for rational targeted therapy. Clin. Cancer Res. 2013, 19, 3796–3807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amary, M.F.; Ye, H.; Forbes, G.; Damato, S.; Maggiani, F.; Pollock, R.; Tirabosco, R.; Flanagan, A.M. Isocitrate dehydrogenase 1 mutations (IDH1) and p16/CDKN2A copy number change in conventional chondrosarcomas. Virchows Arch. 2015, 466, 217–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Fritchie, K.; Wei, S.; Ali, N.; Curless, K.; Shen, T.; Brini, A.T.; Latif, F.; Sumathi, V.; Siegal, G.P.; et al. Diagnostic utility of IDH1/2 mutations to distinguish dedifferentiated chondrosarcoma from undifferentiated pleomorphic sarcoma of bone. Hum. Pathol. 2017, 65, 239–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.; Li, X.S.; Woon, E.C.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Xiao, M.T.; Liu, L.X.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Berzero, G.; Di Stefano, A.L.; Sanson, M. The clinical use of IDH1 and IDH2 mutations in gliomas. Expert Rev. Mol. Diagn. 2018, 18, 1041–1051. [Google Scholar] [CrossRef]
- Hameetman, L.; Rozeman, L.B.; Lombaerts, M.; Oosting, J.; Taminiau, A.H.; Cleton-Jansen, A.M.; Bovee, J.V.; Hogendoorn, P.C. Peripheral chondrosarcoma progression is accompanied by decreased Indian Hedgehog signalling. J. Pathol. 2006, 209, 501–511. [Google Scholar] [CrossRef]
- Addie, R.D.; de Jong, Y.; Alberti, G.; Kruisselbrink, A.B.; Que, I.; Baelde, H.; Bovee, J. Exploration of the chondrosarcoma metabolome; the mTOR pathway as an important pro-survival pathway. J. Bone Oncol. 2019, 15, 100222. [Google Scholar] [CrossRef]
- Schrage, Y.M.; Briaire-de Bruijn, I.H.; de Miranda, N.F.; van Oosterwijk, J.; Taminiau, A.H.; van Wezel, T.; Hogendoorn, P.C.; Bovee, J.V. Kinome profiling of chondrosarcoma reveals SRC-pathway activity and dasatinib as option for treatment. Cancer Res. 2009, 69, 6216–6222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Oosterwijk, J.G.; Meijer, D.; van Ruler, M.A.; van den Akker, B.E.; Oosting, J.; Krenacs, T.; Picci, P.; Flanagan, A.M.; Liegl-Atzwanger, B.; Leithner, A.; et al. Screening for potential targets for therapy in mesenchymal, clear cell, and dedifferentiated chondrosarcoma reveals Bcl-2 family members and TGFbeta as potential targets. Am. J. Pathol. 2013, 182, 1347–1356. [Google Scholar] [CrossRef] [PubMed]
- Van Oosterwijk, J.G.; Herpers, B.; Meijer, D.; Briaire-de Bruijn, I.H.; Cleton-Jansen, A.M.; Gelderblom, H.; van de Water, B.; Bovee, J.V. Restoration of chemosensitivity for doxorubicin and cisplatin in chondrosarcoma in vitro: BCL-2 family members cause chemoresistance. Ann. Oncol. 2012, 23, 1617–1626. [Google Scholar] [CrossRef] [PubMed]
- Bovee, J.V.; Cleton-Jansen, A.M.; Wuyts, W.; Caethoven, G.; Taminiau, A.H.; Bakker, E.; Van Hul, W.; Cornelisse, C.J.; Hogendoorn, P.C. EXT-mutation analysis and loss of heterozygosity in sporadic and hereditary osteochondromas and secondary chondrosarcomas. Am. J. Hum. Genet. 1999, 65, 689–698. [Google Scholar] [CrossRef] [Green Version]
- De Andrea, C.E.; Reijnders, C.M.; Kroon, H.M.; de Jong, D.; Hogendoorn, P.C.; Szuhai, K.; Bovee, J.V. Secondary peripheral chondrosarcoma evolving from osteochondroma as a result of outgrowth of cells with functional EXT. Oncogene 2012, 31, 1095–1104. [Google Scholar] [CrossRef]
- De Andrea, C.E.; Zhu, J.F.; Jin, H.; Bovee, J.V.; Jones, K.B. Cell cycle deregulation and mosaic loss of Ext1 drive peripheral chondrosarcomagenesis in the mouse and reveal an intrinsic cilia deficiency. J. Pathol. 2015, 236, 210–218. [Google Scholar] [CrossRef] [Green Version]
- Fei, L.; Ngoh, C.; Porter, D.E. Chondrosarcoma transformation in hereditary multiple exostoses: A systematic review and clinical and cost-effectiveness of a proposed screening model. J. Bone Oncol. 2018, 13, 114–122. [Google Scholar] [CrossRef]
- McCormick, C.; Leduc, Y.; Martindale, D.; Mattison, K.; Esford, L.E.; Dyer, A.P.; Tufaro, F. The putative tumour suppressor EXT1 alters the expression of cell-surface heparan sulfate. Nat. Genet. 1998, 19, 158–161. [Google Scholar] [CrossRef]
- Hallor, K.H.; Staaf, J.; Bovee, J.V.; Hogendoorn, P.C.; Cleton-Jansen, A.M.; Knuutila, S.; Savola, S.; Niini, T.; Brosjo, O.; Bauer, H.C.; et al. Genomic profiling of chondrosarcoma: Chromosomal patterns in central and peripheral tumors. Clin. Cancer Res. 2009, 15, 2685–2694. [Google Scholar] [CrossRef] [Green Version]
- Cleven, A.H.; Zwartkruis, E.; Hogendoorn, P.C.; Kroon, H.M.; Briaire-de Bruijn, I.; Bovee, J.V. Periosteal chondrosarcoma: A histopathological and molecular analysis of a rare chondrosarcoma subtype. Histopathology 2015, 67, 483–490. [Google Scholar] [CrossRef]
- Nishio, J.; Reith, J.D.; Ogose, A.; Maale, G.; Neff, J.R.; Bridge, J.A. Cytogenetic findings in clear cell chondrosarcoma. Cancer Genet. Cytogenet. 2005, 162, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.K.; Cho, C.H.; Chi, S.G.; Han, C.S.; Ushigome, S.; Unni, K.K. Low incidence of genetic alterations of the p16CDKN2a in clear cell chondrosarcoma. Int. J. Oncol. 2001, 19, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.K.; Park, H.R.; Chi, S.G.; Ushigome, S.; Unni, K.K. Overexpression of p53 and absent genetic mutation in clear cell chondrosarcoma. Int. J. Oncol. 2001, 19, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Behjati, S.; Tarpey, P.S.; Presneau, N.; Scheipl, S.; Pillay, N.; Van Loo, P.; Wedge, D.C.; Cooke, S.L.; Gundem, G.; Davies, H.; et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat. Genet. 2013, 45, 1479–1482. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Motoi, T.; Khanin, R.; Olshen, A.; Mertens, F.; Bridge, J.; Dal Cin, P.; Antonescu, C.R.; Singer, S.; Hameed, M.; et al. Identification of a novel, recurrent HEY1-NCOA2 fusion in mesenchymal chondrosarcoma based on a genome-wide screen of exon-level expression data. Genes Chromosomes Cancer 2012, 51, 127–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyquist, K.B.; Panagopoulos, I.; Thorsen, J.; Haugom, L.; Gorunova, L.; Bjerkehagen, B.; Fossa, A.; Guriby, M.; Nome, T.; Lothe, R.A.; et al. Whole-transcriptome sequencing identifies novel IRF2BP2-CDX1 fusion gene brought about by translocation t(1;5)(q42;q32) in mesenchymal chondrosarcoma. PLoS ONE 2012, 7, e49705. [Google Scholar] [CrossRef]
- Amary, F.; Perez-Casanova, L.; Ye, H.; Cottone, L.; Strobl, A.C.; Cool, P.; Miranda, E.; Berisha, F.; Aston, W.; Rocha, M.; et al. Synovial chondromatosis and soft tissue chondroma: Extraosseous cartilaginous tumor defined by FN1 gene rearrangement. Mod. Pathol. 2019, 32, 1762–1771. [Google Scholar] [CrossRef]
- Delattre, O.; Zucman, J.; Plougastel, B.; Desmaze, C.; Melot, T.; Peter, M.; Kovar, H.; Joubert, I.; de Jong, P.; Rouleau, G.; et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 1992, 359, 162–165. [Google Scholar] [CrossRef]
- Sorensen, P.H.; Lessnick, S.L.; Lopez-Terrada, D.; Liu, X.F.; Triche, T.J.; Denny, C.T. A second Ewing’s sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERG. Nat. Genet. 1994, 6, 146–151. [Google Scholar] [CrossRef]
- Jeon, I.S.; Davis, J.N.; Braun, B.S.; Sublett, J.E.; Roussel, M.F.; Denny, C.T.; Shapiro, D.N. A variant Ewing’s sarcoma translocation (7;22) fuses the EWS gene to the ETS gene ETV1. Oncogene 1995, 10, 1229–1234. [Google Scholar]
- Peter, M.; Couturier, J.; Pacquement, H.; Michon, J.; Thomas, G.; Magdelenat, H.; Delattre, O. A new member of the ETS family fused to EWS in Ewing tumors. Oncogene 1997, 14, 1159–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, T.L.; O’Sullivan, M.J.; Pallen, C.J.; Hayes, M.; Clarkson, P.W.; Winstanley, M.; Sorensen, P.H.; Nielsen, T.O.; Horsman, D.E. Ewing sarcoma with novel translocation t(2;16) producing an in-frame fusion of FUS and FEV. J. Mol. Diagn. 2007, 9, 459–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorensen, P.H.; Liu, X.F.; Delattre, O.; Rowland, J.M.; Biggs, C.A.; Thomas, G.; Triche, T.J. Reverse transcriptase PCR amplification of EWS/FLI-1 fusion transcripts as a diagnostic test for peripheral primitive neuroectodermal tumors of childhood. Diagn. Mol. Pathol. 1993, 2, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Walsh, M.F.; Wu, G.; Edmonson, M.N.; Gruber, T.A.; Easton, J.; Hedges, D.; Ma, X.; Zhou, X.; Yergeau, D.A.; et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N. Engl. J. Med. 2015, 373, 2336–2346. [Google Scholar] [CrossRef] [Green Version]
- Rahman, N. Realizing the promise of cancer predisposition genes. Nature 2014, 505, 302–308. [Google Scholar] [CrossRef] [Green Version]
- Grunewald, T.G.; Bernard, V.; Gilardi-Hebenstreit, P.; Raynal, V.; Surdez, D.; Aynaud, M.M.; Mirabeau, O.; Cidre-Aranaz, F.; Tirode, F.; Zaidi, S.; et al. Chimeric EWSR1-FLI1 regulates the Ewing sarcoma susceptibility gene EGR2 via a GGAA microsatellite. Nat. Genet. 2015, 47, 1073–1078. [Google Scholar] [CrossRef] [Green Version]
- Tomazou, E.M.; Sheffield, N.C.; Schmidl, C.; Schuster, M.; Schonegger, A.; Datlinger, P.; Kubicek, S.; Bock, C.; Kovar, H. Epigenome mapping reveals distinct modes of gene regulation and widespread enhancer reprogramming by the oncogenic fusion protein EWS-FLI1. Cell Rep. 2015, 10, 1082–1095. [Google Scholar] [CrossRef] [Green Version]
- Beck, R.; Monument, M.J.; Watkins, W.S.; Smith, R.; Boucher, K.M.; Schiffman, J.D.; Jorde, L.B.; Randall, R.L.; Lessnick, S.L. EWS/FLI-responsive GGAA microsatellites exhibit polymorphic differences between European and African populations. Cancer Genet. 2012, 205, 304–312. [Google Scholar] [CrossRef] [Green Version]
- Grunewald, T.G.P.; Cidre-Aranaz, F.; Surdez, D.; Tomazou, E.M.; de Alava, E.; Kovar, H.; Sorensen, P.H.; Delattre, O.; Dirksen, U. Ewing sarcoma. Nat. Rev. Dis. Primers 2018, 4, 5. [Google Scholar] [CrossRef]
- Brohl, A.S.; Solomon, D.A.; Chang, W.; Wang, J.; Song, Y.; Sindiri, S.; Patidar, R.; Hurd, L.; Chen, L.; Shern, J.F.; et al. The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet. 2014, 10, e1004475. [Google Scholar] [CrossRef] [Green Version]
- Toretsky, J.A.; Kalebic, T.; Blakesley, V.; LeRoith, D.; Helman, L.J. The insulin-like growth factor-I receptor is required for EWS/FLI-1 transformation of fibroblasts. J. Biol. Chem. 1997, 272, 30822–30827. [Google Scholar] [CrossRef] [Green Version]
- Rocchi, A.; Manara, M.C.; Sciandra, M.; Zambelli, D.; Nardi, F.; Nicoletti, G.; Garofalo, C.; Meschini, S.; Astolfi, A.; Colombo, M.P.; et al. CD99 inhibits neural differentiation of human Ewing sarcoma cells and thereby contributes to oncogenesis. J. Clin. Investig. 2010, 120, 668–680. [Google Scholar] [CrossRef] [PubMed]
- Franzetti, G.A.; Laud-Duval, K.; van der Ent, W.; Brisac, A.; Irondelle, M.; Aubert, S.; Dirksen, U.; Bouvier, C.; de Pinieux, G.; Snaar-Jagalska, E.; et al. Cell-to-cell heterogeneity of EWSR1-FLI1 activity determines proliferation/migration choices in Ewing sarcoma cells. Oncogene 2017, 36, 3505–3514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werner, H.; Meisel-Sharon, S.; Bruchim, I. Oncogenic fusion proteins adopt the insulin-like growth factor signaling pathway. Mol. Cancer 2018, 17, 28. [Google Scholar] [CrossRef] [PubMed]
- Yee, D.; Favoni, R.E.; Lebovic, G.S.; Lombana, F.; Powell, D.R.; Reynolds, C.P.; Rosen, N. Insulin-like growth factor I expression by tumors of neuroectodermal origin with the t(11;22) chromosomal translocation. A potential autocrine growth factor. J. Clin. Investig. 1990, 86, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Scotlandi, K.; Benini, S.; Sarti, M.; Serra, M.; Lollini, P.L.; Maurici, D.; Picci, P.; Manara, M.C.; Baldini, N. Insulin-like growth factor I receptor-mediated circuit in Ewing’s sarcoma/peripheral neuroectodermal tumor: A possible therapeutic target. Cancer Res. 1996, 56, 4570–4574. [Google Scholar]
- Benini, S.; Manara, M.C.; Baldini, N.; Cerisano, V.; Massimo, S.; Mercuri, M.; Lollini, P.L.; Nanni, P.; Picci, P.; Scotlandi, K. Inhibition of insulin-like growth factor I receptor increases the antitumor activity of doxorubicin and vincristine against Ewing’s sarcoma cells. Clin. Cancer Res. 2001, 7, 1790–1797. [Google Scholar]
- Olmos, D.; Postel-Vinay, S.; Molife, L.R.; Okuno, S.H.; Schuetze, S.M.; Paccagnella, M.L.; Batzel, G.N.; Yin, D.; Pritchard-Jones, K.; Judson, I.; et al. Safety, pharmacokinetics, and preliminary activity of the anti-IGF-1R antibody figitumumab (CP-751,871) in patients with sarcoma and Ewing’s sarcoma: A phase 1 expansion cohort study. Lancet Oncol. 2010, 11, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Juergens, H.; Daw, N.C.; Geoerger, B.; Ferrari, S.; Villarroel, M.; Aerts, I.; Whelan, J.; Dirksen, U.; Hixon, M.L.; Yin, D.; et al. Preliminary efficacy of the anti-insulin-like growth factor type 1 receptor antibody figitumumab in patients with refractory Ewing sarcoma. J. Clin. Oncol. 2011, 29, 4534–4540. [Google Scholar] [CrossRef] [Green Version]
- Pappo, A.S.; Patel, S.R.; Crowley, J.; Reinke, D.K.; Kuenkele, K.P.; Chawla, S.P.; Toner, G.C.; Maki, R.G.; Meyers, P.A.; Chugh, R.; et al. R1507, a monoclonal antibody to the insulin-like growth factor 1 receptor, in patients with recurrent or refractory Ewing sarcoma family of tumors: Results of a phase II Sarcoma Alliance for Research through Collaboration study. J. Clin. Oncol. 2011, 29, 4541–4547. [Google Scholar] [CrossRef]
- Tap, W.D.; Demetri, G.; Barnette, P.; Desai, J.; Kavan, P.; Tozer, R.; Benedetto, P.W.; Friberg, G.; Deng, H.; McCaffery, I.; et al. Phase II study of ganitumab, a fully human anti-type-1 insulin-like growth factor receptor antibody, in patients with metastatic Ewing family tumors or desmoplastic small round cell tumors. J. Clin. Oncol. 2012, 30, 1849–1856. [Google Scholar] [CrossRef] [PubMed]
- Mancarella, C.; Scotlandi, K. IGF system in sarcomas: A crucial pathway with many unknowns to exploit for therapy. J. Mol. Endocrinol. 2018, 61, T45–T60. [Google Scholar] [CrossRef] [PubMed]
- Mancarella, C.; Pasello, M.; Manara, M.C.; Toracchio, L.; Sciandra, E.F.; Picci, P.; Scotlandi, K. Insulin-Like Growth Factor 2 mRNA-Binding Protein 3 Influences Sensitivity to Anti-IGF System Agents Through the Translational Regulation of IGF1R. Front. Endocrinol. 2018, 9, 178. [Google Scholar] [CrossRef] [PubMed]
- Gellini, M.; Ascione, A.; Flego, M.; Mallano, A.; Dupuis, M.L.; Zamboni, S.; Terrinoni, M.; D’Alessio, V.; Manara, M.C.; Scotlandi, K.; et al. Generation of human single-chain antibody to the CD99 cell surface determinant specifically recognizing Ewing’s sarcoma tumor cells. Curr. Pharm. Biotechnol. 2013, 14, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Guerzoni, C.; Fiori, V.; Terracciano, M.; Manara, M.C.; Moricoli, D.; Pasello, M.; Sciandra, M.; Nicoletti, G.; Gellini, M.; Dominici, S.; et al. CD99 triggering in Ewing sarcoma delivers a lethal signal through p53 pathway reactivation and cooperates with doxorubicin. Clin. Cancer Res. 2015, 21, 146–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scotlandi, K.; Perdichizzi, S.; Bernard, G.; Nicoletti, G.; Nanni, P.; Lollini, P.L.; Curti, A.; Manara, M.C.; Benini, S.; Bernard, A.; et al. Targeting CD99 in association with doxorubicin: An effective combined treatment for Ewing’s sarcoma. Eur. J. Cancer 2006, 42, 91–96. [Google Scholar] [CrossRef]
- Manara, M.C.; Pasello, M.; Scotlandi, K. CD99: A Cell Surface Protein with an Oncojanus Role in Tumors. Genes 2018, 9, 159. [Google Scholar] [CrossRef] [Green Version]
- Pasello, M.; Manara, M.C.; Scotlandi, K. CD99 at the crossroads of physiology and pathology. J. Cell Commun. Signal. 2018, 12, 55–68. [Google Scholar] [CrossRef] [Green Version]
- Celik, H.; Sciandra, M.; Flashner, B.; Gelmez, E.; Kayraklioglu, N.; Allegakoen, D.V.; Petro, J.R.; Conn, E.J.; Hour, S.; Han, J.; et al. Clofarabine inhibits Ewing sarcoma growth through a novel molecular mechanism involving direct binding to CD99. Oncogene 2018, 37, 2181–2196. [Google Scholar] [CrossRef]
- Crompton, B.D.; Stewart, C.; Taylor-Weiner, A.; Alexe, G.; Kurek, K.C.; Calicchio, M.L.; Kiezun, A.; Carter, S.L.; Shukla, S.A.; Mehta, S.S.; et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014, 4, 1326–1341. [Google Scholar] [CrossRef] [Green Version]
- Tirode, F.; Surdez, D.; Ma, X.; Parker, M.; Le Deley, M.C.; Bahrami, A.; Zhang, Z.; Lapouble, E.; Grossetete-Lalami, S.; Rusch, M.; et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov. 2014, 4, 1342–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Vire, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; Van Eynde, A.; Bernard, D.; Vanderwinden, J.M.; et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Dominguez, D.J.; Hontecillas-Prieto, L.; Rodriguez-Nunez, P.; Pascual-Pasto, G.; Vila-Ubach, M.; Garcia-Mejias, R.; Robles, M.J.; Tirado, O.M.; Mora, J.; Carcaboso, A.M.; et al. The combination of epigenetic drugs SAHA and HCI-2509 synergistically inhibits EWS-FLI1 and tumor growth in Ewing sarcoma. Oncotarget 2018, 9, 31397–31410. [Google Scholar] [CrossRef] [Green Version]
- Erkizan, H.V.; Kong, Y.; Merchant, M.; Schlottmann, S.; Barber-Rotenberg, J.S.; Yuan, L.; Abaan, O.D.; Chou, T.H.; Dakshanamurthy, S.; Brown, M.L.; et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma. Nat. Med. 2009, 15, 750–756. [Google Scholar] [CrossRef] [Green Version]
- Spriano, F.; Chung, E.Y.L.; Gaudio, E.; Tarantelli, C.; Cascione, L.; Napoli, S.; Jessen, K.; Carrassa, L.; Priebe, V.; Sartori, G.; et al. The ETS Inhibitors YK-4-279 and TK-216 Are Novel Antilymphoma Agents. Clin. Cancer Res. 2019, 25, 5167–5176. [Google Scholar] [CrossRef]
- Kennedy, A.L.; Vallurupalli, M.; Chen, L.; Crompton, B.; Cowley, G.; Vazquez, F.; Weir, B.A.; Tsherniak, A.; Parasuraman, S.; Kim, S.; et al. Functional, chemical genomic, and super-enhancer screening identify sensitivity to cyclin D1/CDK4 pathway inhibition in Ewing sarcoma. Oncotarget 2015, 6, 30178–30193. [Google Scholar] [CrossRef] [Green Version]
- Brenner, J.C.; Feng, F.Y.; Han, S.; Patel, S.; Goyal, S.V.; Bou-Maroun, L.M.; Liu, M.; Lonigro, R.; Prensner, J.R.; Tomlins, S.A.; et al. PARP-1 inhibition as a targeted strategy to treat Ewing’s sarcoma. Cancer Res. 2012, 72, 1608–1613. [Google Scholar] [CrossRef] [Green Version]
- Gorthi, A.; Romero, J.C.; Loranc, E.; Cao, L.; Lawrence, L.A.; Goodale, E.; Iniguez, A.B.; Bernard, X.; Masamsetti, V.P.; Roston, S.; et al. EWS-FLI1 increases transcription to cause R-loops and block BRCA1 repair in Ewing sarcoma. Nature 2018, 555, 387–391. [Google Scholar] [CrossRef]
- Choy, E.; Butrynski, J.E.; Harmon, D.C.; Morgan, J.A.; George, S.; Wagner, A.J.; D’Adamo, D.; Cote, G.M.; Flamand, Y.; Benes, C.H.; et al. Phase II study of olaparib in patients with refractory Ewing sarcoma following failure of standard chemotherapy. BMC Cancer 2014, 14, 813. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, P.; Likhotvorik, R.; Baig, N.; Cropper, J.; Carlson, R.; Kurmasheva, R.; Sridhar, S. Nanoformulation of Talazoparib Increases Maximum Tolerated Doses in Combination With Temozolomide for Treatment of Ewing Sarcoma. Front. Oncol. 2019, 9, 1416. [Google Scholar] [CrossRef] [PubMed]
- Schafer, E.S.; Rau, R.E.; Berg, S.L.; Liu, X.; Minard, C.G.; Bishop, A.J.R.; Romero, J.C.; Hicks, M.J.; Nelson, M.D., Jr.; Voss, S.; et al. Phase 1/2 trial of talazoparib in combination with temozolomide in children and adolescents with refractory/recurrent solid tumors including Ewing sarcoma: A Children’s Oncology Group Phase 1 Consortium study (ADVL1411). Pediatr. Blood Cancer 2020, 67, e28073. [Google Scholar] [CrossRef] [PubMed]
- Heske, C.M.; Davis, M.I.; Baumgart, J.T.; Wilson, K.; Gormally, M.V.; Chen, L.; Zhang, X.; Ceribelli, M.; Duveau, D.Y.; Guha, R.; et al. Matrix Screen Identifies Synergistic Combination of PARP Inhibitors and Nicotinamide Phosphoribosyltransferase (NAMPT) Inhibitors in Ewing Sarcoma. Clin. Cancer Res. 2017, 23, 7301–7311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.A.; Reynolds, C.P.; Kang, M.H.; Kolb, E.A.; Gorlick, R.; Carol, H.; Lock, R.B.; Keir, S.T.; Maris, J.M.; Billups, C.A.; et al. Synergistic activity of PARP inhibition by talazoparib (BMN 673) with temozolomide in pediatric cancer models in the pediatric preclinical testing program. Clin. Cancer Res. 2015, 21, 819–832. [Google Scholar] [CrossRef] [Green Version]
- Salvador-Barbero, B.; Alvarez-Fernandez, M.; Zapatero-Solana, E.; El Bakkali, A.; Menendez, M.D.C.; Lopez-Casas, P.P.; Di Domenico, T.; Xie, T.; VanArsdale, T.; Shields, D.J.; et al. CDK4/6 Inhibitors Impair Recovery from Cytotoxic Chemotherapy in Pancreatic Adenocarcinoma. Cancer Cell 2020, 37, 340–353. [Google Scholar] [CrossRef]
Figure 1.
Transition toward personalized therapies in high-grade osteosarcoma.

Figure 2.
Role of isocitrate dehydrogenase (IDH) and IDH-mutated forms on the inhibition of demethylation pathways and its possible impact on chondrosarcoma genesis. Instead of isocitrate being converted to α-ketoglutarate (α-KG) with the production of reduced nicotinamide adenine dinucleotide phosphate (NADPH), α-KG is converted to 2-hydroxyglutarate (2-HG) with the consumption of reduced nicotinamide adenine dinucleotide phosphate NADPH. 2-HG is expressed at high levels in tumor cells and supports tumorigenesis by modulating Jumonji-C (JmjC)-mediated histone methylation and Tet methylcytosine dioxygenase TET-mediated DNA methylation. ROS, reactive oxygen species.
Figure 2.
Role of isocitrate dehydrogenase (IDH) and IDH-mutated forms on the inhibition of demethylation pathways and its possible impact on chondrosarcoma genesis. Instead of isocitrate being converted to α-ketoglutarate (α-KG) with the production of reduced nicotinamide adenine dinucleotide phosphate (NADPH), α-KG is converted to 2-hydroxyglutarate (2-HG) with the consumption of reduced nicotinamide adenine dinucleotide phosphate NADPH. 2-HG is expressed at high levels in tumor cells and supports tumorigenesis by modulating Jumonji-C (JmjC)-mediated histone methylation and Tet methylcytosine dioxygenase TET-mediated DNA methylation. ROS, reactive oxygen species.


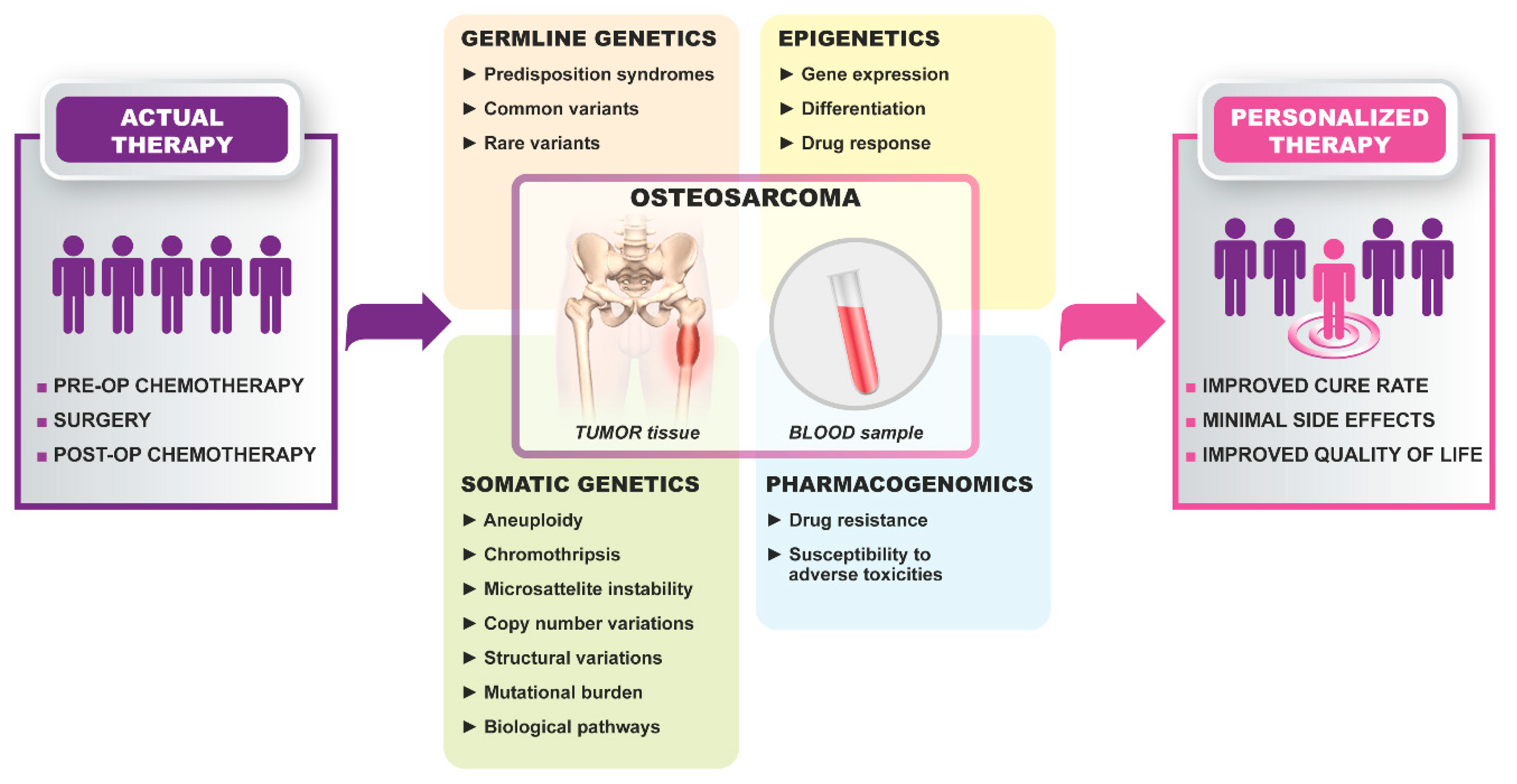{kind=link}
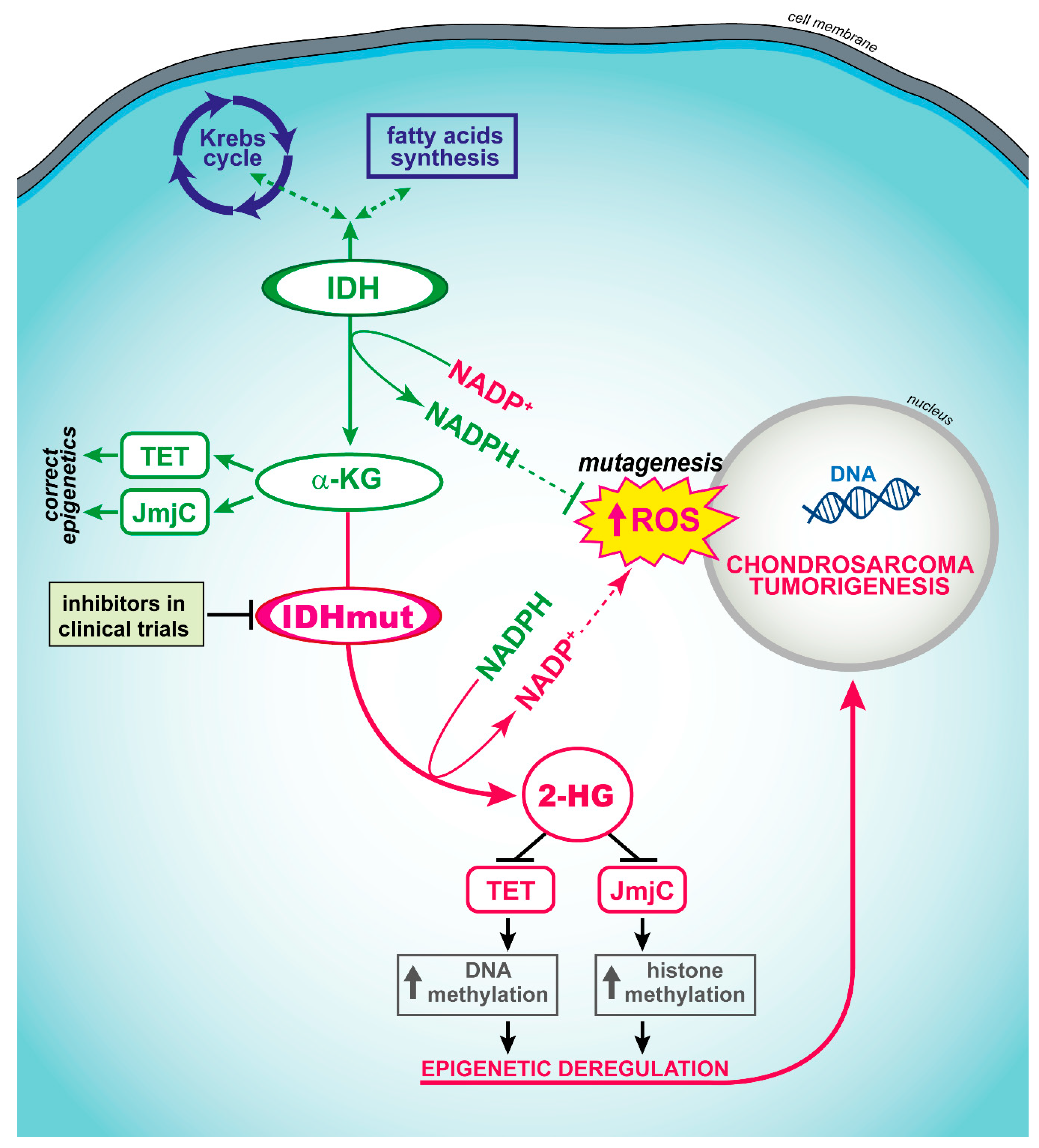{kind=link}
Table 1.
List of target-specific clinical trials that are presently active and recruiting high-grade osteosarcoma (HGOS) patients. Time period refers to the actual study start date and estimated study completion date.
Table 1.
List of target-specific clinical trials that are presently active and recruiting high-grade osteosarcoma (HGOS) patients. Time period refers to the actual study start date and estimated study completion date.
| Treatment | Mechanism of Action | Bone Sarcoma Histotypes | ClinicalTrials.gov NCT Identifier (Protocol Acronym) | Participating Countries | Stage of Development (Time Period) |
|---|---|---|---|---|---|
| Losartan + Sunitinib | Sunitinib: multi-target inhibition of RTK | HGOS | NCT03900793 | USA | phase I (08/2019–02/2025) |
| Famitinib plus Camrelizumab (SHR-1210) or Famitinib alone or Famitinib plus Ifosfamide | Famitinib: multi-target inhibition of RTK, including SCFR (c-Kit), VEGFR2 and 3, PDGFR, Flt1 and Flt3 Camrelizumab: inhibition of PD-1 immune checkpoint | advanced HGOS | NCT04044378 | China | phase I/II (08/2019–09/2022) |
| Pazopanib hydrochloride (Votrient®) with oral Topotecan hydrochloride | Inhibition of VEGFR-1, -2, -3, PDGFR-α and -β, and KIT (Pazopanib) | recurrent or metastatic HGOS | NCT02357810 | USA | phase II (02/2015–06/2022) |
| Apatinib (YN968D1) in combination with chemotherapy | Inhibition of VEGFR2 | HGOS with pulmonary metastasis | NCT03742193 | China | phase II (03/2019–09/2022) |
| Regorafenib (BAY 73-4506, commercial name Stivarga) | Multi-kinase inhibitor targeting VEGFR2, TIE2, PDGFR-beta, FGFR, KIT, RET, and RAF | HGOS, Ewing sarcoma | NCT02048371 (SARC024) | USA | phase II (07/2014–12/2020) |
| Regorafenib (BAY 73-4506, commercial name Stivarga) | Multi-kinase inhibitor targeting VEGFR2, TIE2, PDGFR-beta, FGFR, KIT, RET, and RAF | metastatic bone sarcomas (HGOS, Ewing sarcoma, chondrosarcoma) | NCT02389244 (REGOBONE) | France | phase II (09/2014–03/2023) |
| Cabozantinib-S-Malate (Cabometyx; Cometriq) | Inhibition of MET, VEGFR2, AXL and RET | recurrent, refractory, or newly diagnosed sarcomas, including HGOS | NCT02867592 | USA | phase II (05/2017–06/2020) |
| Erdafitinib (JNJ-42756493) | FGFR inhibitor with negative effects on angiogenesis | relapsed or refractory advanced solid tumors, including HGOS | NCT03210714 (The Pediatric MATCH Screening Trial) | USA | phase II (11/2017–12/2024) |
| Palbociclib (PD-0332991, trade name Ibrance) | Selective inhibition of the cyclin-dependent kinases CDK4 and CDK6 | relapsed or refractory advanced solid tumors, including refractory HGOS | NCT03526250 (The Pediatric MATCH Screening Trial) | USA | phase II (06/2018–06/2025) |
| Abemaciclib (Verzenios) | Selective inhibition of the cyclin-dependent kinases CDK4 and CDK6 | recurrent or refractory solid tumors, including HGOS and Ewing sarcoma | NCT02644460 | USA | phase I (02/2016–12/2020) |
| Abemaciclib (Verzenios) | Selective inhibition of the cyclin-dependent kinases CDK4 and CDK6 | advanced HGOS and chondrosarcoma | NCT04040205 | USA | phase II (10/2019–09/2024) |
| Samotolisib (LY3023414) | Inhibition of PI3K/AKT/mTOR pathway | relapsed or refractory advanced solid tumors, including HGOS | NCT03213678 (The Pediatric MATCH Screening Trial) | USA | phase II (07/2017–09/2024) |
| Berzosertib (M6620; VX-970; Captisol®) | Selective inhibitor of ATR | HGOS | NCT03718091 | USA | phase II (01/2019–04/2025) |
| Olaparib (AZD-2281, MK-7339 trade name Lynparza®) | Inhibition of PARP1, opposing DNA repair, in patients with hereditary BRCA1 or BRCA2 mutations | relapsed or refractory advanced solid tumors, including HGOS | NCT03233204 (The Pediatric MATCH Screening Trial) | USA | phase II (07/2017–09/2024) |
| Ulixertinib (BVD-523; VRT752271) | Inhibition of ERK1/2 kinases, belonging to the MAPK pathway | relapsed or refractory advanced solid tumors, including recurrent HGOS | NCT03698994 (The Pediatric MATCH Screening Trial) | USA | phase II (10/2018–12/2025) |
| Vemurafenib (PLX40321; Zelboraf®) | Inhibition of the mutated B-Raf protein, interrupting its stimulation of cell growth | relapsed or refractory advanced solid tumors, including HGOS | NCT03220035 (The Pediatric MATCH Screening Trial) | USA | phase II (07/2017–12/2023) |
| Larotrectinib (ARRY-470; LOXO-101; Vitrakvi®) | Inhibition of tropomyosin kinase receptors TrkA, TrkB, and TrkC | relapsed or refractory advanced solid tumors, including HGOS | NCT03213704 | USA | phase II (07/2017–09/2024) |
| 9-ING-41 with Gemcitabine, Doxorubicin, Lomustine, Carboplatin, Nab paclitaxel, Paclitaxel | 9-ING-41: inhibition of GSK-3 | advanced cancers, including HGOS | NCT03678883 | USA | phase I/II (01/2019–11/2022) |
| Tazemetostat (EPZ-6438) | Inhibition of the activity of human polycomb repressive complex 2 -containing wild-type histone-lysine N-methyltransferases EZH1 and EZH2 | advanced cancers, including HGOS and Ewing sarcoma | NCT03213665 (The Pediatric MATCH Screening Trial) | USA | phase II (07/2017–09/2024) |
| Avelumab (Bavencio®) | Targeting PD-L1 | recurrent or progressive HGOS | NCT03006848 | USA | phase II (02/2017–01/2023) |
| ZKAB001 (STI-1014; STI-A1014) | Targeting PD-L1 | recurrent or refractory HGOS | NCT03676985 | China | phase I/II (10/2018–06/2023) |
| Nivolumab (Opdivo®) with or without Azacitidine | Targeting PD-1 (Nivolumab) | recurrent HGOS | NCT03628209 | USA | phase I/II (07/2019–07/2022) |
| Pembrolizumab (MK3475) combined with metronomic Cyclophosphamide | Targeting PD-1 in association with metronomic chemotherapy | advanced sarcomas | NCT02406781 | France | phase II (06/2015–06/2023) |
| Pepinemab (VX15/2503) | Targeting Semaphorin-4D (SEMA4D), also known as Cluster of Differentiation 100 (CD100), which binds to CD72 to activate B cells and dendritic cells | recurrent and refractory HGOS | NCT03320330 | USA | phase I/II (01/2018–09/2021) |
| 4th generation safety-engineered CAR T cells targeting sarcomas | 4th generation safety-engineered CAR T cells targeting sarcoma surface antigens | sarcomas including HGOS and Ewing sarcoma | NCT03356782 | China | phase I (12/2017–12/2020) |
| EGFR806 CAR T cell immunotherapy | second generation EGFR-specific CAR T cells, which have been genetically modified to express either the EGFR receptor alone (EGFR806CAR(2G)-EGFRt) or in addition also the CD19 receptor (CD19CAR(2G)-T2A-HER2tG) | recurrent or refractory solid tumors, including HGOS and Ewing sarcoma | NCT03618381 | USA | phase I (06/2019–06/2036) |
| C7R-GD2.CAR T cell immunotherapy | relapsed or refractory GD2-positive tumors, including HGOS and Ewing sarcoma | NCT03635632 | USA | phase I (04/2019–12/2037) | |
| Humanized Monoclonal Antibody 3F8 (Hu3F8) combined with GM-CSF | Targeting GD2 with the humanized antibody Hu3F8 | recurrent HGOS | NCT02502786 | USA | phase II (07/2015–07/2021) |
| Humanized anti-GD2 bispecific antibody Hu3F8-BsAb | Targeting GD2 with the bispecific antibody Hu3F8-BsAb | HGOS | NCT03860207 | USA | phase I/II (02/2019–02/2022) |
Legend: ATR, ataxia telangiectasia and rad3-related; BRCA, breast related cancer antigen; CD100, cluster of differentiation 100; CDK, cyclin-dependent kinase; EGFR epidermal growth factor receptor; ERK, extracellular signal–regulated kinase; Flt, FMS-like tyrosine kinase; FGFR, fibroblast growth factor receptor; GD2, disialoganglioside 2; GSK-3, glycogen sinthase kinase 3; HGOS, high-grade osteosarcoma; mTOR, mammalian target of rapamycin; MAPK, mitogen-activated protein kinase; PDGFR, platelet-derived growth factor receptor; PARP1, poly (ADP-ribose) polymerase 1; PI3K, phosphatidylinositol 3-kinase; RAF, rapidly accelerated fibrosarcoma; PD-1, programmed cell death 1; PD-L1, programmed death-ligand 1; SCFR (c-Kit) stem cell factor receptor; RET, rearranged during transfection; RTK, receptor tyrosine kinase; SEMA4D, semaphorin-4D; TRK, tropomyosin kinase receptor; TIE2, tyrosine kinase with immunoglobulin-like and EGF-like domains 2; VEGFR, vascular endothelial growth factor receptor.
Table 2.
List of target-specific clinical trials that are presently active and recruiting chondrosarcoma patients. Trials that are also recruiting high-grade osteosarcoma patients are listed in Table 1. Time period refer to the actual study start date and estimated study completion date.
Table 2.
List of target-specific clinical trials that are presently active and recruiting chondrosarcoma patients. Trials that are also recruiting high-grade osteosarcoma patients are listed in Table 1. Time period refer to the actual study start date and estimated study completion date.
| Treatment | Mechanism of Action | Bone Sarcoma Histotypes | ClinicalTrials.gov NCT Identifier (Protocol Acronym) | Participating Countries | Stage of Development (Time Period) |
|---|---|---|---|---|---|
| Sirolimus and Cyclophosphamide | Inhibition of mTOR signalling | Conventional, Mesenchymal and Dedifferentiated Chondrosarcomas | NCT02821507 (COSYMO) | Netherlands Spain | phase II (06/2014–06/2021) |
| FT 2102 (Olutasidenib) | Inhibitor of mutant IDH1 | Chondrosarcoma | NCT03684811 | USA | phase II (11/2018–04/2022) |
Legend: IDH1, Isocitrate dehydrogenase 1; mTOR, mammalian target of rapamycin.
Table 3.
List of target-specific clinical trials that are presently active and recruiting Ewing sarcoma patients. Trials that are also recruiting high-grade osteosarcoma or chondrosarcoma patients are listed in Table 1 and Table 2. Time period refers to the actual study start date and estimated study completion date.
Table 3.
List of target-specific clinical trials that are presently active and recruiting Ewing sarcoma patients. Trials that are also recruiting high-grade osteosarcoma or chondrosarcoma patients are listed in Table 1 and Table 2. Time period refers to the actual study start date and estimated study completion date.
| Treatment | Mechanism of Action | Bone Sarcoma Histotypes | ClinicalTrials.gov NCT Identifier (Protocol Acronym) | Participating Countries | Stage of Development (Time Period) |
|---|---|---|---|---|---|
| SP-2577 (Seclidemstat) | Inhibition of the LSD1 epigenetic enzyme | Ewing Sarcoma | NCT03600649 | USA | phase I (06/2018–12/2021) |
| INCB059872 | Inhibition of the LSD1 epigenetic enzyme | relapsed or refractory Ewing sarcoma | NCT03514407 | USA Italy Spain UK | phase I (06/2018–06/2021) |
| Niraparib and Temozolomide and/or Irinotecan | Niraparib: inhibition of PARP | previously treated, incurable Ewing sarcoma | NCT02044120 | USA UK | phase I (05/2014–04/2021) |
| Pbi-shRNA™ EWS/FLI1 Type 1 LPX | Targeting EWS/FLI1 type 1 fusion transcript | advanced Ewing sarcoma | NCT02736565 | USA | phase I (10/2016–02/2022) |
| TK216 | Inhibition of the between EWS-FLI1 and RNA helicase A through binding to EWS-FLI1 | relapsed or refractory Ewing sarcoma | NCT02657005 | USA | phase I (08/2016–06/2021) |
| Vorinostat (Zolinza) in combination with chemotherapy | Inhibition of HDAC | Ewing Sarcoma | NCT04308330 | USA | phase I (03/2017–12/2022) |
| Olaparib and Temozolomide | Inhibition of PARP | Ewing Sarcoma | NCT01858168 | USA | phase I (07/2013–07/2024) |
| Anlotinib and Irinotecan | Multi-target inhibition of RTK, including VEGFR2 and VEGFR3 | metastatic Ewing Sarcoma | NCT03416517 | China | phase I (01/2018–12/2020) |
| Palbociclib combined with chemotherapy | Inhibition of CDK4/6 | Ewing Sarcoma | NCT03709680 | USA | phase I (05/2019–04/2024) |
| Palbociclib plus Ganitumab | Palbociclib: inhibition of CDK4/6 Ganitumab: inhibition of IGF-1R | Ewing Sarcoma | NCT04129151 | USA | phase II (12/2019–08/2022) |
| Eribulin Mesylate | Inhibition polymerization of tubulin subunits impairing the EWS-FLI1 mediated microtubule stabilization | Ewing Sarcoma | NCT03441360 | USA | phase II (04/2018–06/2020) |
| Eribulin Mesylate | Inhibition polymerization of tubulin subunits impairing the EWS-FLI1 mediated microtubule stabilization | Ewing Sarcoma | NCT03245450 | France Germany Greece Italy Spain UK | phase II (08/2017–09/2021) |
| Nivolumab (Opdivo®) plus ABI-009 (Nab-rapamycin; Nab-sirolimus) | Targeting PD-1 (Nivolumab) and mTOR (ABI-009) | Ewing sarcoma | NCT03190174 | USA | phase I/II (08/2017–04/2021) |
Legend: CDK, cyclin dependent kinase; EWS-FLI1, Ewing Sarcoma-Friend Leukemia Insertion; HDAC: histone deacetylases; IGF-1, Insulin-like growth factor 1 receptor; LSD1, lysine-specific demethylase 1 enzyme; mTOR, mammalian target of rapamycin; PARP, poly (ADP-ribose) polymerase; PD-1, programmed cell death 1; RTK, receptor tyrosine kinase; VEGFR, vascular endothelial growth factor receptor.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Scotlandi, K.; Hattinger, C.M.; Pellegrini, E.; Gambarotti, M.; Serra, M. Genomics and Therapeutic Vulnerabilities of Primary Bone Tumors. Cells 2020, 9, 968. https://doi.org/10.3390/cells9040968
AMA Style
Scotlandi K, Hattinger CM, Pellegrini E, Gambarotti M, Serra M. Genomics and Therapeutic Vulnerabilities of Primary Bone Tumors. Cells. 2020; 9(4):968. https://doi.org/10.3390/cells9040968
Chicago/Turabian StyleScotlandi, Katia, Claudia Maria Hattinger, Evelin Pellegrini, Marco Gambarotti, and Massimo Serra. 2020. "Genomics and Therapeutic Vulnerabilities of Primary Bone Tumors" Cells 9, no. 4: 968. https://doi.org/10.3390/cells9040968
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.