

The Mitochondrial Connection: The Nek Kinases’ New Functional Axis in Mitochondrial Homeostasis

 ,
,
Abstract
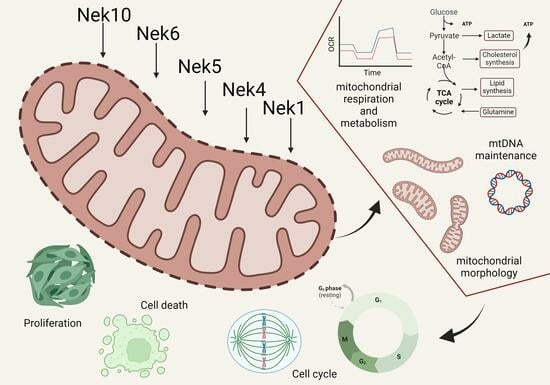
:
1. Introduction
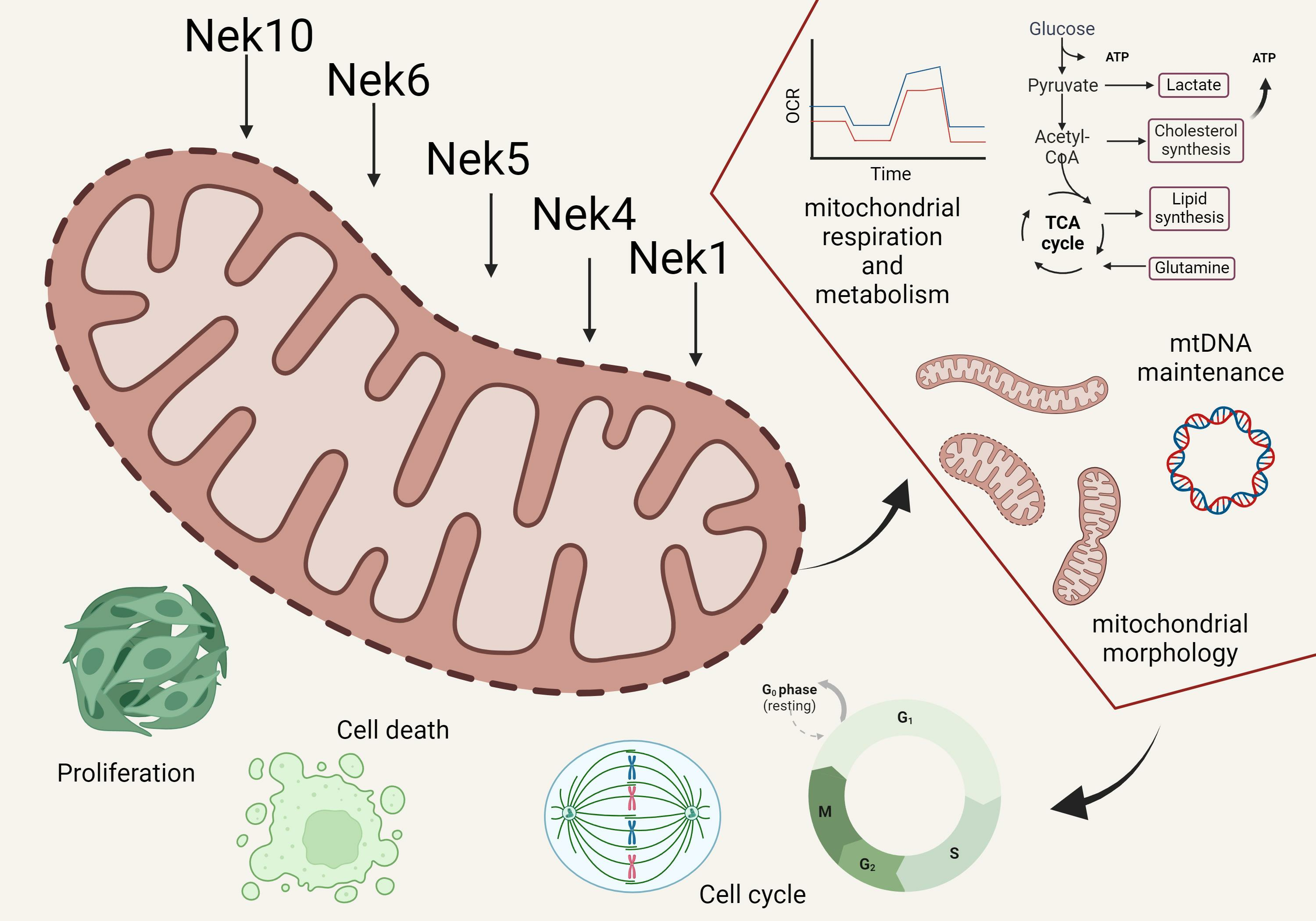
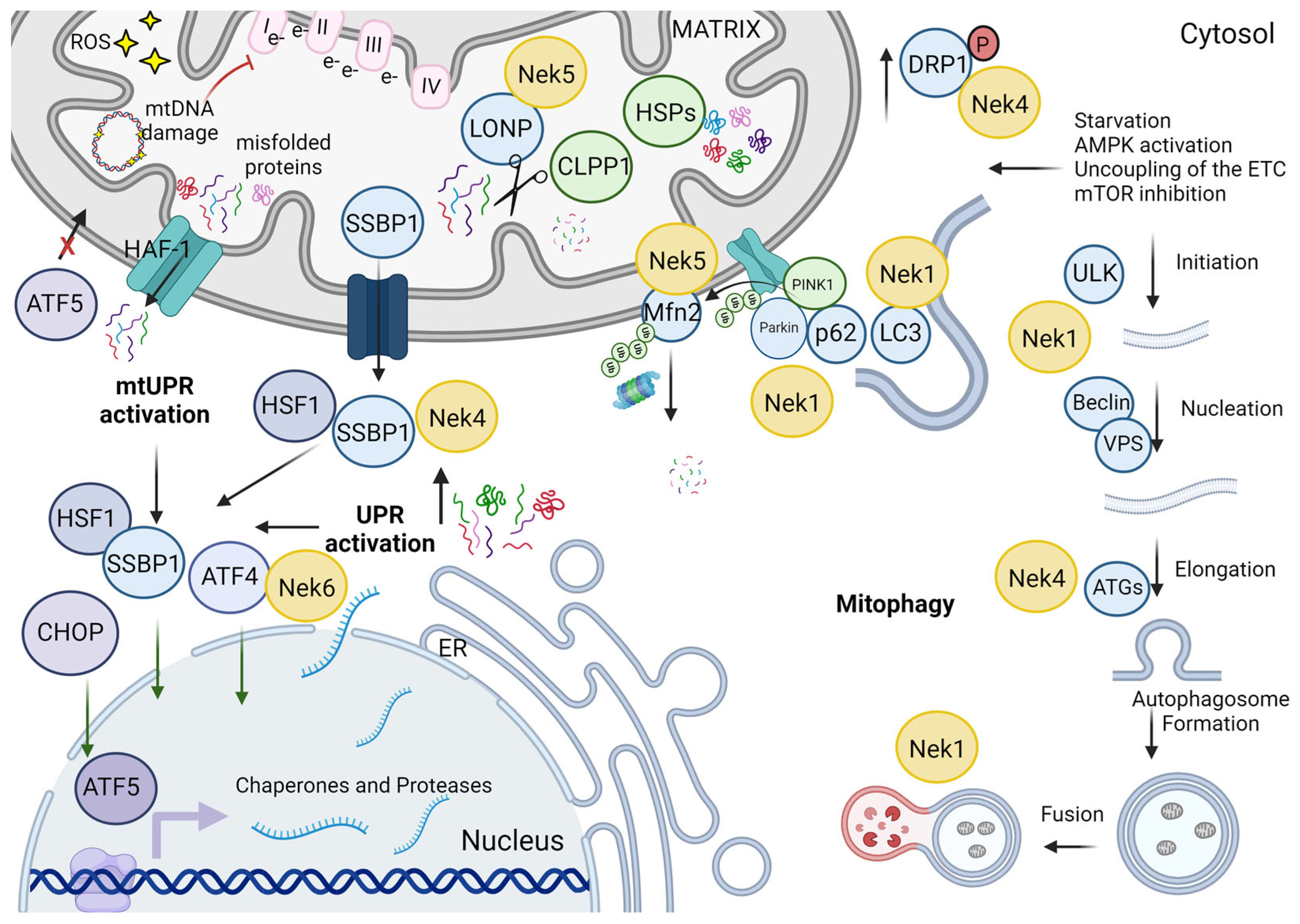
2. Nek Interactomes Reveal Mitochondrion-Related Functions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biological Process (BP, Mitochondrial Related Genes) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Autophagy | Apoptosis | Stress Response | Respiration-Related | |||||
| % of Genes | p-Value | % of Genes | p-Value | % of Genes | p-Value | % of Genes | p-Value | |
| Nek1 | 4.3 | 1.3 × 10−5 | 8.1 | 1.5 × 10−4 | 6.2 | 9.6 × 10−3 | - | - |
| Nek4 | - | - | - | - | 1.9 | 3.0 × 10−3 | 0.6 | 6.5 × 10−2 |
| Nek5 | 3.5 | 2.2 × 10−3 | - | - | 3.5 | 1.2 × 10−4 | - | - |
| Nek10 | - | - | - | - | - | - | 13 | 7.3 × 10−3 |
| MitoProt II | Target 2.0 | MitoFates | ||
|---|---|---|---|---|
| Probability | mTP | Probability of Presequence | TOMM20 Recognition Motif | |
| Nek1 | 0.0356 | 0.000163 | 0.003 | Yes |
| Nek2A | 0.1593 | 0.000069 | 0.002 | Yes |
| Nek2B | 0.1367 | 0.000069 | 0.002 | Yes |
| Nek2C | 0.2477 | 0.000069 | 0.002 | Yes |
| Nek3 | 0.0518 | 0.001566 | 0.005 | Yes |
| Nek4.1 | 0.1438 | 0.019433 | 0.013 | Yes |
| Nek4.2 | 0.0548 | 0.019433 | 0.013 | Yes |
| Nek5 | 0.1493 | 0.000059 | 0.001 | Yes |
| Nek6 | 0.0055 | 0.000293 | 0.01 | No |
| Nek7 | 0.0044 | 0.000016 | 0 | No |
| Nek8 | 0.1201 | 0.000873 | 0.075 | Yes |
| Nek9 | 0.0242 | 0.000003 | 0 | No |
| Nek10 | 0.0040 | 0.000013 | 0.001 | No |
| Nek11 | 0.7487 | 0.153774 | 0.888 | Yes |
| Mfn1 | 0.2037 | 0.000086 | 0.02 | Yes |
| OPA1 | 0.9981 | 0.915521 | 0.509 | Yes |
| TFAM | 0.9190 | 0.964329 | 0.996 | Yes |
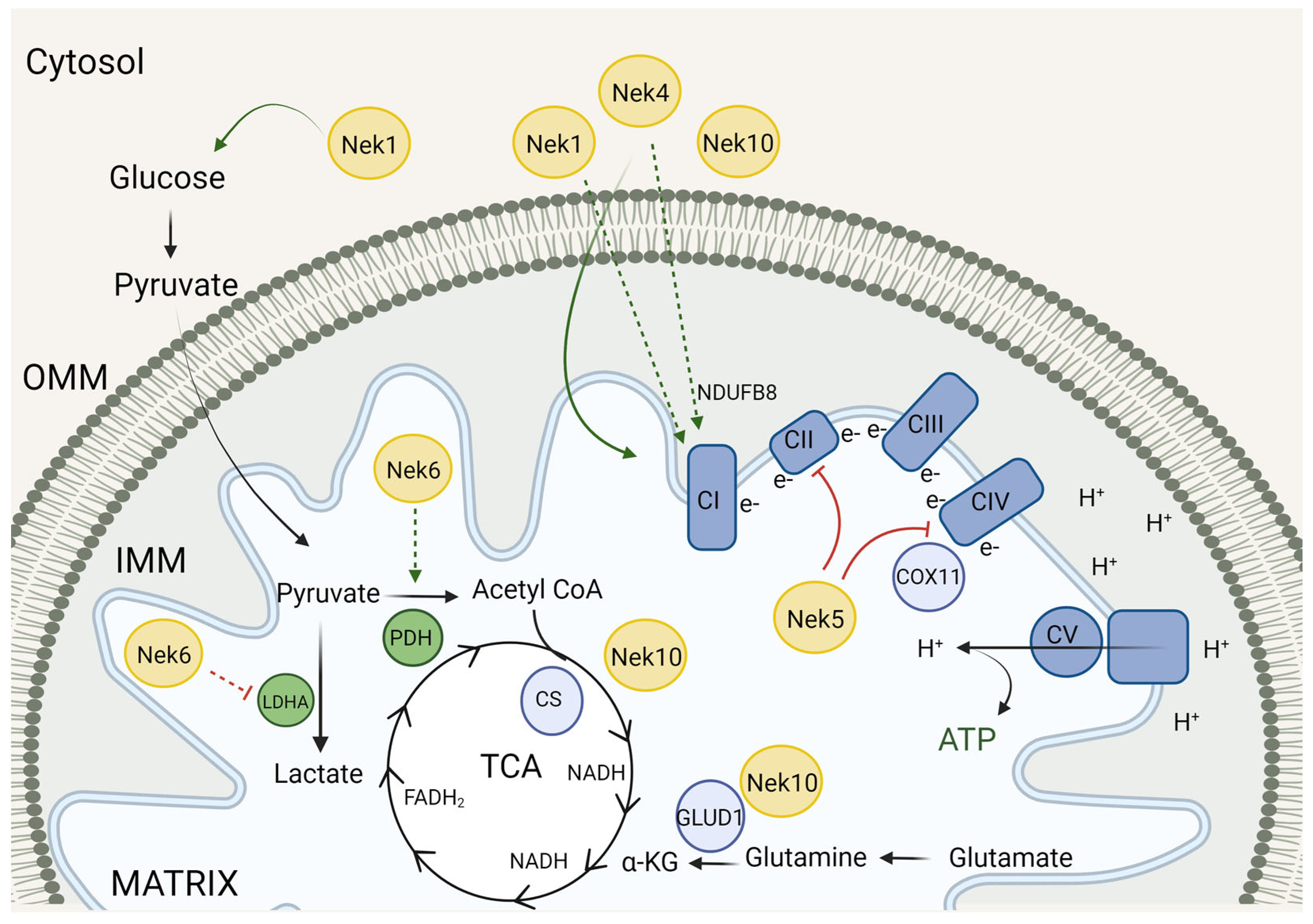
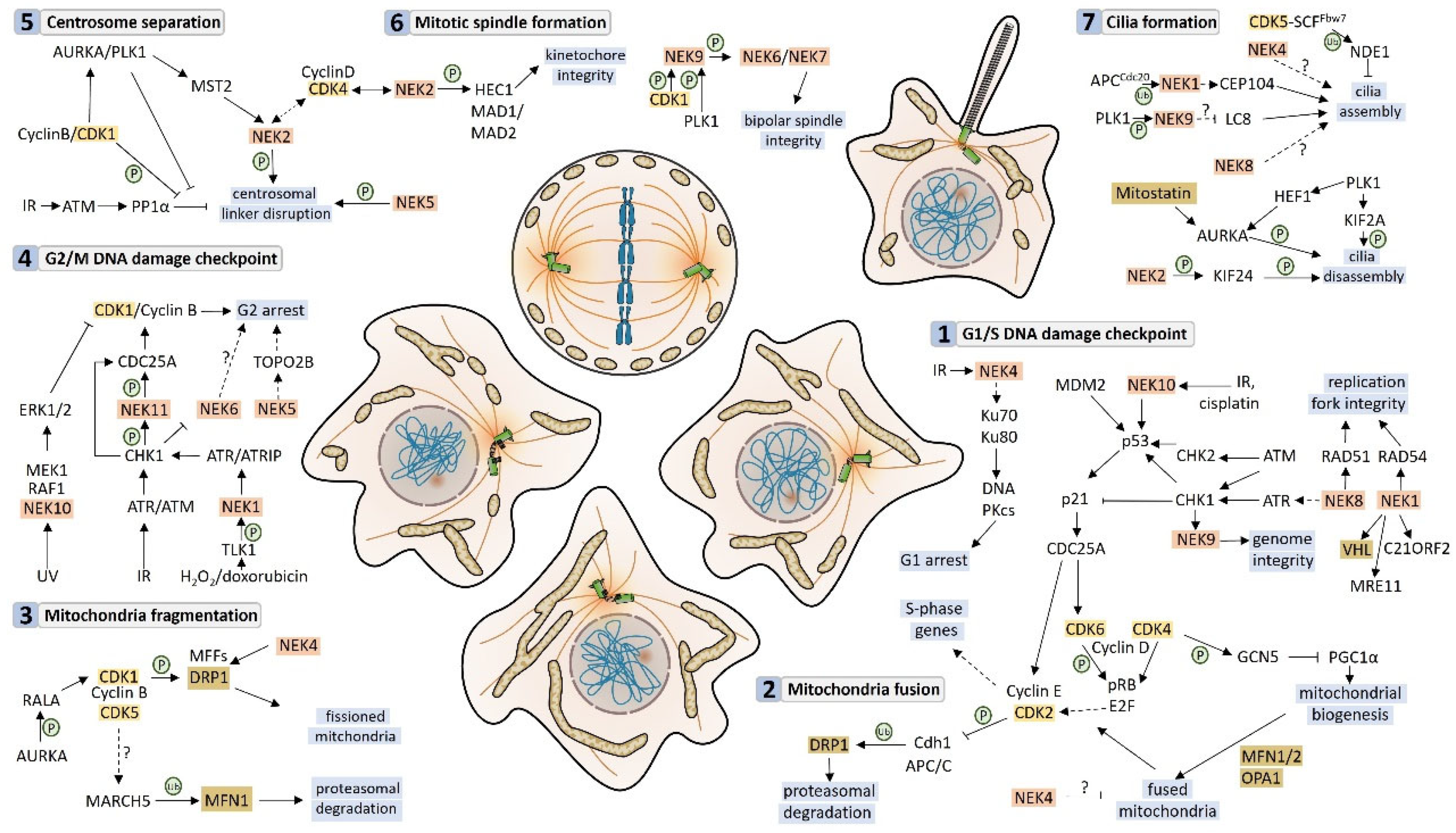
3. Neks and the Mitochondrial Metabolism
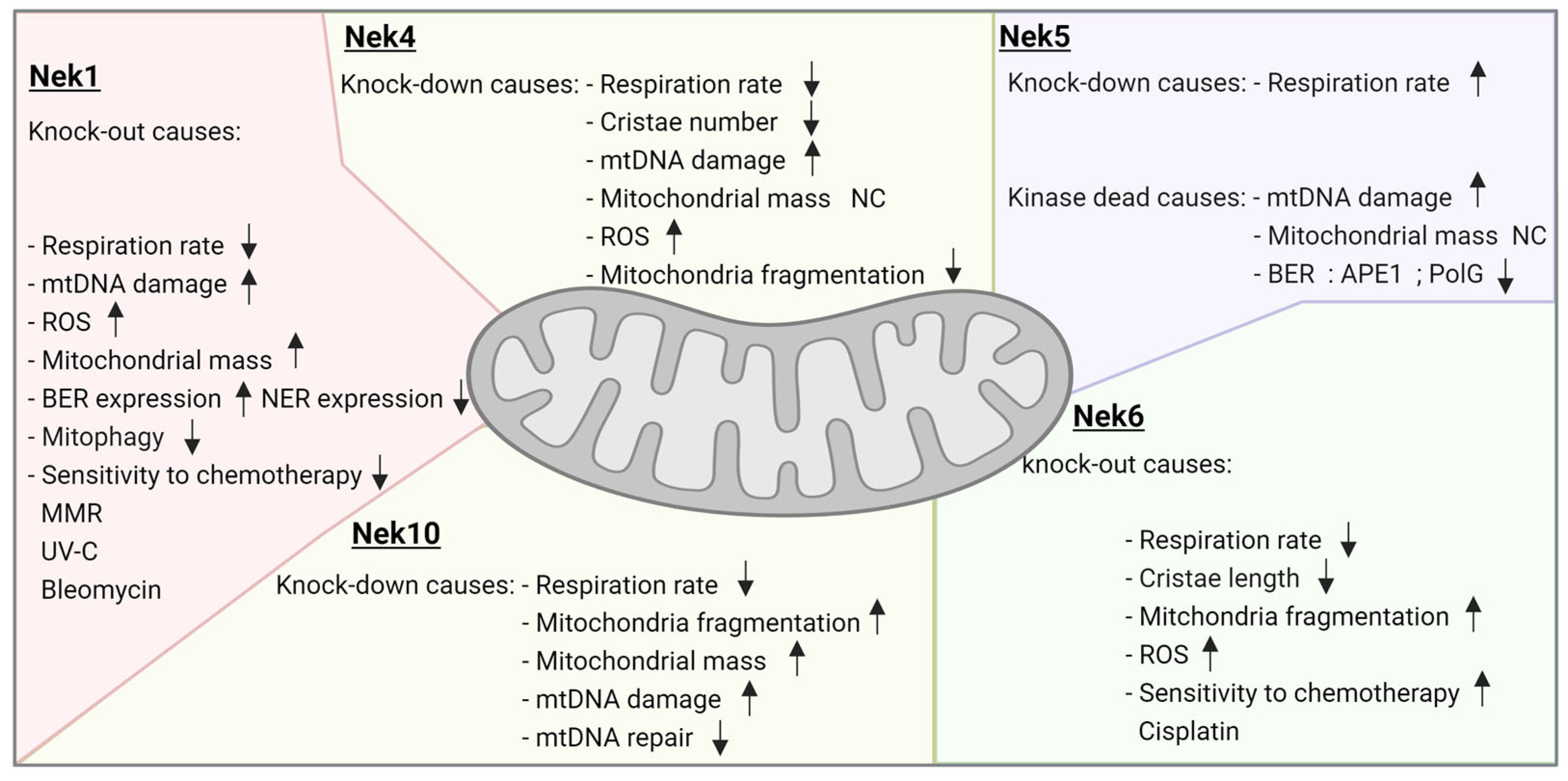
3.1. Neks 1, 4, 6, and 10 Increase Mitochondrial Respiration, as Opposed to Nek5
3.2. Mitochondrial Respiration and Metabolism Partners
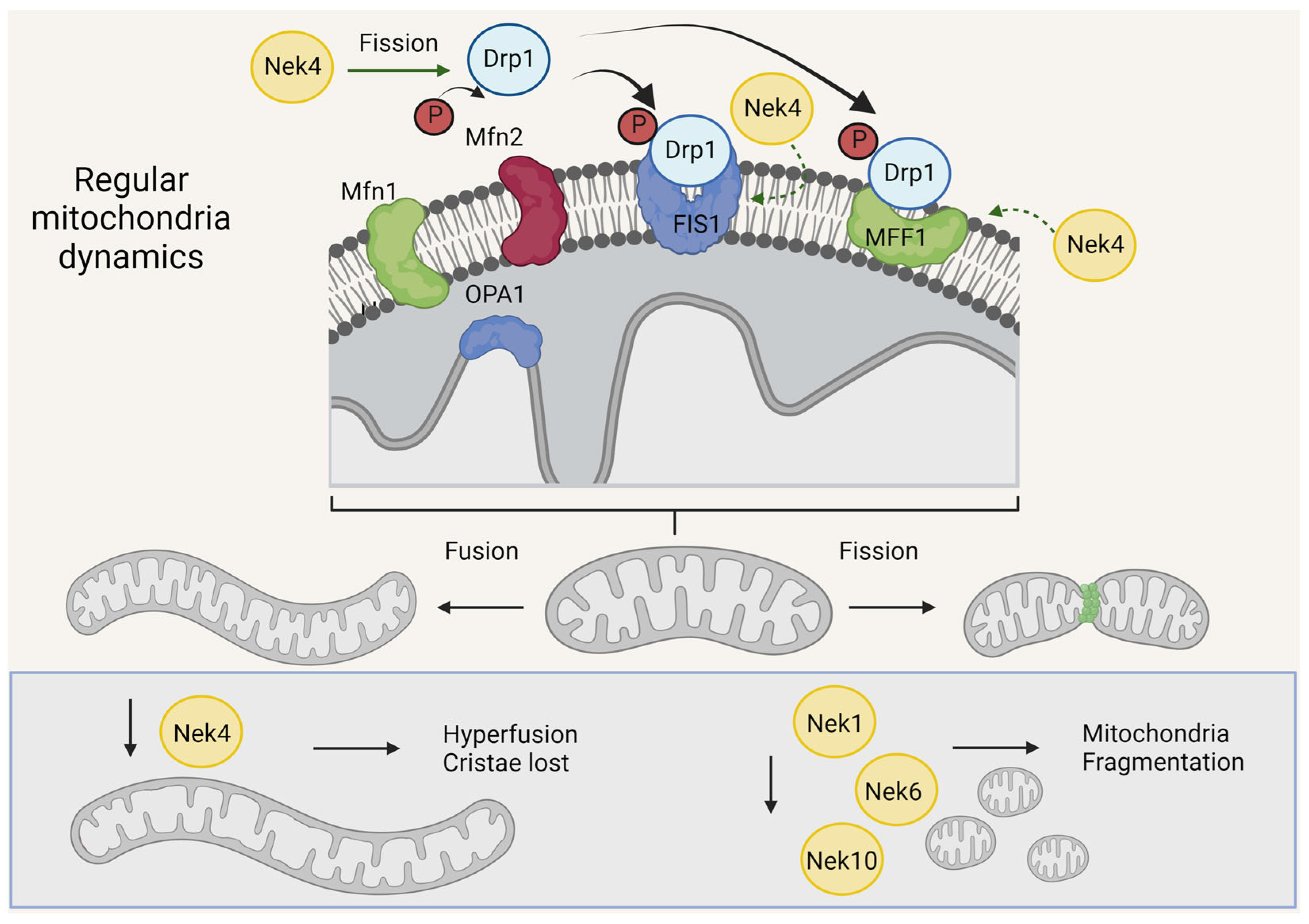
4. Neks and Mitochondrial Morphology
4.1. Nek1, Nek6, and Nek10 Depletion Are Associated with Mitochondrial Fragmentation, as Opposed to Nek4
4.2. Nek Interactors Related to Mitochondrial Morphology
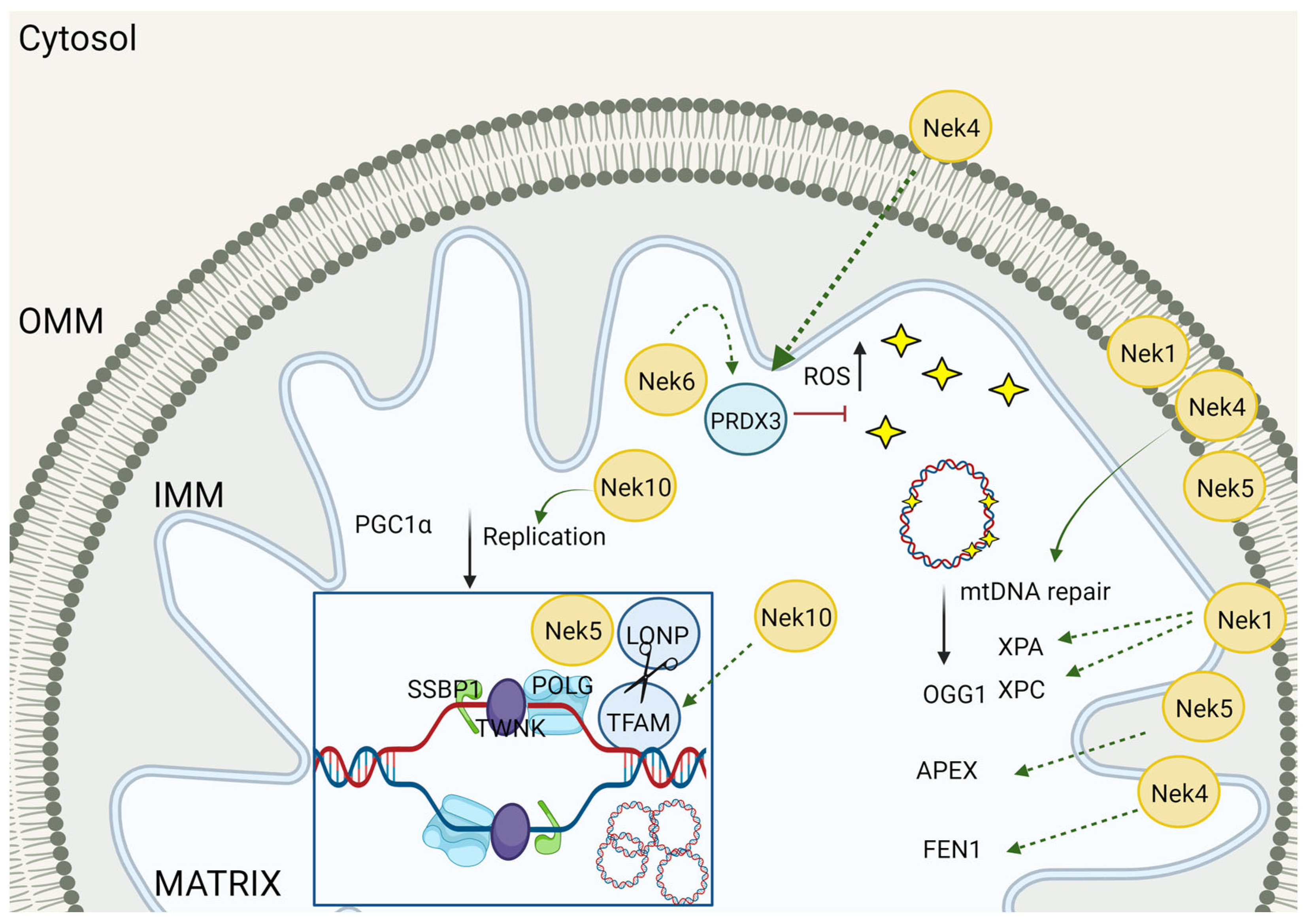
5. mtDNA Maintenance
5.1. Nek Involvement in mtDNA Maintenance
5.2. Neks and the Interaction with DNA Repair-Related Enzymes

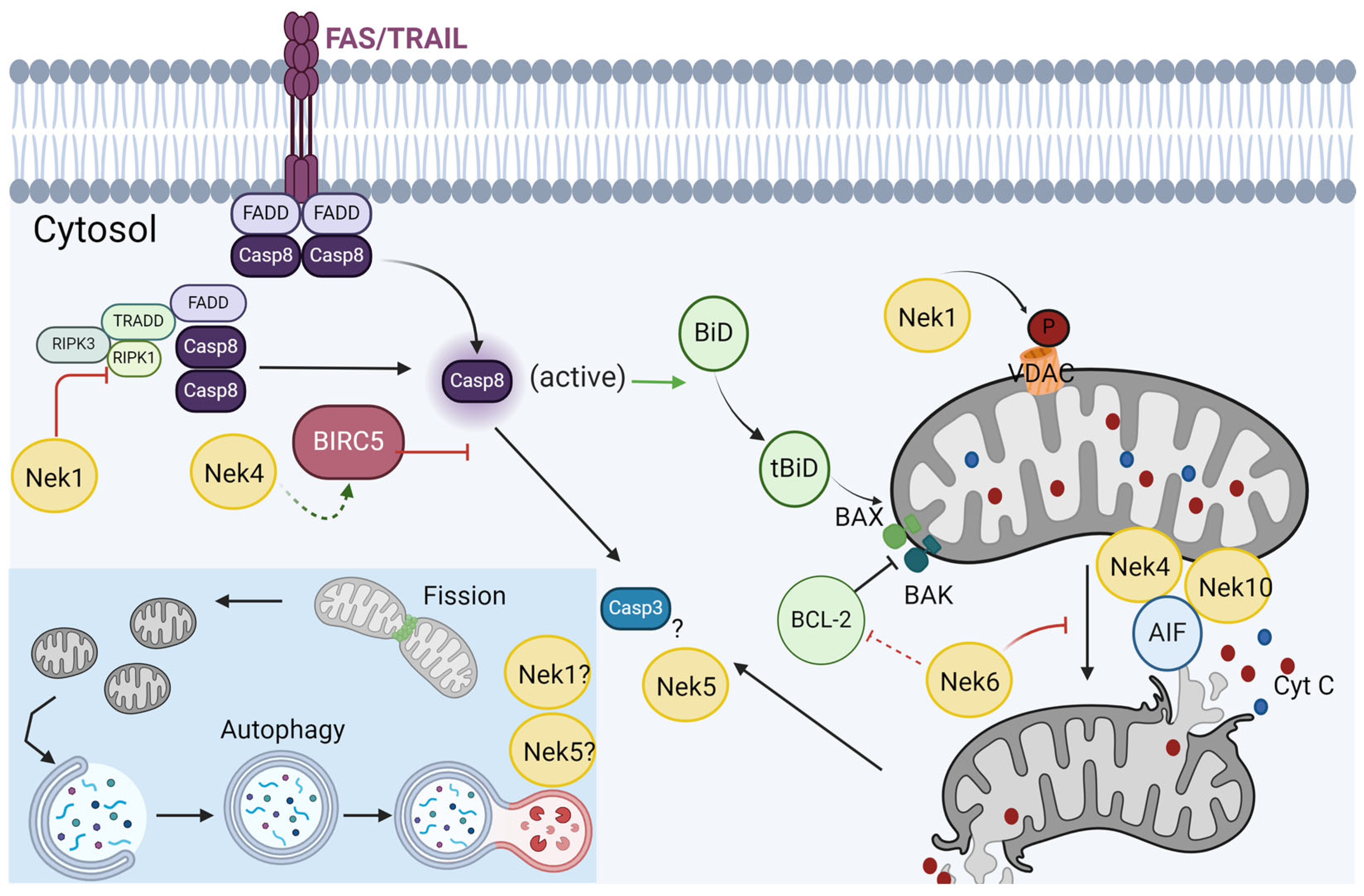
6. Response to Stress and Cell Death
Nek1 and Nek5 Participate in Cell Death in Opposite Ways
7. Response to Stress and Autophagy
8. Mitochondrial versus Cell Cycle-Related Functions
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oakley, B.; Morris, R. A Mutation in Aspergillus Nidulans That Blocks the Transition from Interphase to Prophase. J. Cell Biol. 1983, 96, 1155–1158. [Google Scholar] [CrossRef] [PubMed]
- Osmani, S.A.; May, G.S.; Morris, N.R. Regulation of the mRNA Levels of nimA, a Gene Required for the G2-M Transition in Aspergillus Nidulans. J. Cell Biol. 1987, 104, 1495–1504. [Google Scholar] [CrossRef] [PubMed]
- Morris, N.R.; Enos, A.P. Mitotic Gold in a Mold: Aspergillus Genetics and the Biology of Mitosis. Trends Genet. 1992, 8, 32–33. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M.; Okano, Y. Molecular Cloning and Characterization of the Human NIMA-Related Protein Kinase 3 Gene (NEK3). Cytogenet. Genome Res. 2001, 95, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, K.; Fukazawa, H.; Murakami, Y.; Uehara, Y. Nek11, a New Member of the NIMA Family of Kinases, Involved in DNA Replication and Genotoxic Stress Responses. J. Biol. Chem. 2002, 277, 39655–39665. [Google Scholar] [CrossRef] [PubMed]
- Roig, J.; Mikhailov, A.; Belham, C.; Avruch, J. Nercc1, a Mammalian NIMA-Family Kinase, Binds the Ran GTPase and Regulates Mitotic Progression. Genes Dev. 2002, 16, 1640–1658. [Google Scholar] [CrossRef] [PubMed]
- Feige, E.; Shalom, O.; Tsuriel, S.; Yissachar, N.; Motro, B. Nek1 Shares Structural and Functional Similarities with NIMA Kinase. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2006, 1763, 272–281. [Google Scholar] [CrossRef]
- Moniz, L.S.; Stambolic, V. Nek10 Mediates G2/M Cell Cycle Arrest and MEK Autoactivation in Response to UV Irradiation. Mol. Cell. Biol. 2011, 31, 30–42. [Google Scholar] [CrossRef]
- Meirelles, G.V.; Perez, A.M.; de Souza, E.E.; Basei, F.L.; Papa, P.F.; Melo Hanchuk, T.D.; Cardoso, V.B.; Kobarg, J. “Stop Ne(c)King around”: How Interactomics Contributes to Functionally Characterize Nek Family Kinases. World J. Biol. Chem. 2014, 5, 141–160. [Google Scholar]
- O’Connell, M.J.; Krien, M.J.E.; Hunter, T. Never Say Never. The NIMA-Related Protein Kinases in Mitotic Control. Trends Cell Biol. 2003, 13, 221–228. [Google Scholar] [CrossRef]
- Pavan, I.C.B.; Peres de Oliveira, A.; Dias, P.R.F.; Basei, F.L.; Issayama, L.K.; Ferezin, C.d.C.; Silva, F.R.; Rodrigues de Oliveira, A.L.; Alves dos Reis Moura, L.; Martins, M.B.; et al. On Broken Ne(c)Ks and Broken DNA: The Role of Human NEKs in the DNA Damage Response. Cells 2021, 10, 507. [Google Scholar] [CrossRef] [PubMed]
- Naro, C.; Barbagallo, F.; Chieffi, P.; Bourgeois, C.F.; Paronetto, M.P.; Sette, C. The Centrosomal Kinase NEK2 Is a Novel Splicing Factor Kinase Involved in Cell Survival. Nucleic Acids Res. 2014, 42, 3218–3227. [Google Scholar] [CrossRef] [PubMed]
- Basei, F.L.; Meirelles, G.V.; Righetto, G.L.; dos Santos Migueleti, D.L.; Smetana, J.H.C.; Kobarg, J. New Interaction Partners for Nek4.1 and Nek4.2 Isoforms: From the DNA Damage Response to RNA Splicing. Proteome Sci. 2015, 13, 11. [Google Scholar] [CrossRef]
- Chen, Y.; Craigen, W.J.; Riley, D.J. Nek1 Regulates Cell Death and Mitochondrial Membrane Permeability through Phosphorylation of VDAC1. Cell Cycle 2009, 8, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Gaczynska, M.; Osmulski, P.; Polci, R.; Riley, D.J. Phosphorylation by Nek1 Regulates Opening and Closing of Voltage Dependent Anion Channel 1. Biochem. Biophys. Res. Commun. 2010, 394, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Melo Hanchuk, T.D.; Papa, P.F.; La Guardia, P.G.; Vercesi, A.E.; Kobarg, J. Nek5 Interacts with Mitochondrial Proteins and Interferes Negatively in Mitochondrial Mediated Cell Death and Respiration. Cell. Signal. 2015, 27, 1168–1177. [Google Scholar] [CrossRef]
- Ferezin, C.d.C.; Basei, F.L.; Melo-Hanchuk, T.D.; Oliveira, A.L.; Peres de Oliveira, A.; Mori, M.P.; Souza-Pinto, N.C.; Kobarg, J. NEK5 Interacts with LonP1 and Its Kinase Activity Is Essential for the Regulation of Mitochondrial Functions and mtDNA Maintenance. FEBS Open Bio 2021, 11, 546–563. [Google Scholar] [CrossRef]
- de Castro Ferezin, C.; Lim Kam Sian, T.C.C.; Wu, Y.; Ma, X.; Chüeh, A.C.; Huang, C.; Schittenhelm, R.B.; Kobarg, J.; Daly, R.J. Identification of Biological Pathways and Processes Regulated by NEK5 in Breast Epithelial Cells via an Integrated Proteomic Approach. Cell Commun. Signal. 2022, 20, 197. [Google Scholar] [CrossRef]
- Peres de Oliveira, A.; Basei, F.L.; Slepicka, P.F.; de Castro Ferezin, C.; Melo-Hanchuk, T.D.; de Souza, E.E.; Lima, T.I.; dos Santos, V.T.; Mendes, D.; Silveira, L.R.; et al. NEK10 Interactome and Depletion Reveal New Roles in Mitochondria. Proteome Sci. 2020, 18, 4. [Google Scholar] [CrossRef]
- Basei, F.L.; de Castro Ferezin, C.; Rodrigues de Oliveira, A.L.; Muñoz, J.P.; Zorzano, A.; Kobarg, J. Nek4 Regulates Mitochondrial Respiration and Morphology. FEBS J. 2022, 289, 3262–3279. [Google Scholar] [CrossRef]
- da Silva, F.R.; UNICAMP, Campinas, SP, Brazil. Personal communication, 2024.
- Melo-Hanchuk, T.D.; Slepicka, P.F.; Meirelles, G.V.; Basei, F.L.; Lovato, D.V.; Granato, D.C.; Pauletti, B.A.; Domingues, R.R.; Leme, A.F.P.; Pelegrini, A.L.; et al. NEK1 Kinase Domain Structure and Its Dynamic Protein Interactome after Exposure to Cisplatin. Sci. Rep. 2017, 7, 5445. [Google Scholar] [CrossRef] [PubMed]
- Vaz Meirelles, G.; Ferreira Lanza, D.C.; da Silva, J.C.; Santana Bernachi, J.; Paes Leme, A.F.; Kobarg, J. Characterization of hNek6 Interactome Reveals an Important Role for Its Short N-Terminal Domain and Colocalization with Proteins at the Centrosome. J. Proteome Res. 2010, 9, 6298–6316. [Google Scholar] [CrossRef] [PubMed]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A Web Server for Functional Enrichment Analysis and Functional Annotation of Gene Lists (2021 Update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef] [PubMed]
- Bolender, N.; Sickmann, A.; Wagner, R.; Meisinger, C.; Pfanner, N. Multiple Pathways for Sorting Mitochondrial Precursor Proteins. EMBO Rep. 2008, 9, 42–49. [Google Scholar] [CrossRef]
- Dudek, J.; Rehling, P.; van der Laan, M. Mitochondrial Protein Import: Common Principles and Physiological Networks. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2013, 1833, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, N.; Pfanner, N. Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem. 2017, 86, 685–714. [Google Scholar] [CrossRef]
- Jores, T.; Klinger, A.; Groß, L.E.; Kawano, S.; Flinner, N.; Duchardt-Ferner, E.; Wöhnert, J.; Kalbacher, H.; Endo, T.; Schleiff, E.; et al. Characterization of the Targeting Signal in Mitochondrial β-Barrel Proteins. Nat. Commun. 2016, 7, 12036. [Google Scholar] [CrossRef]
- Bykov, Y.S.; Flohr, T.; Boos, F.; Zung, N.; Herrmann, J.M.; Schuldiner, M. Widespread Use of Unconventional Targeting Signals in Mitochondrial Ribosome Proteins. EMBO J. 2022, 41, e109519. [Google Scholar] [CrossRef]
- Lee, C.M.; Sedman, J.; Neupert, W.; Stuart, R.A. The DNA Helicase, Hmi1p, Is Transported into Mitochondria by a C-Terminal Cleavable Targeting Signal. J. Biol. Chem. 1999, 274, 20937–20942. [Google Scholar] [CrossRef]
- Backes, S.; Bykov, Y.S.; Flohr, T.; Räschle, M.; Zhou, J.; Lenhard, S.; Krämer, L.; Mühlhaus, T.; Bibi, C.; Jann, C.; et al. The Chaperone-Binding Activity of the Mitochondrial Surface Receptor Tom70 Protects the Cytosol against Mitoprotein-Induced Stress. Cell Rep. 2021, 35, 108936. [Google Scholar] [CrossRef]
- Almagro Armenteros, J.J.; Salvatore, M.; Emanuelsson, O.; Winther, O.; von Heijne, G.; Elofsson, A.; Nielsen, H. Detecting Sequence Signals in Targeting Peptides Using Deep Learning. Life Sci. Alliance 2019, 2, e201900429. [Google Scholar] [CrossRef] [PubMed]
- Claros, M.G.; Vincens, P. Computational Method to Predict Mitochondrially Imported Proteins and Their Targeting Sequences. Eur. J. Biochem. 1996, 241, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, N.; Kozjak, V.; Chacinska, A.; Schönfisch, B.; Rospert, S.; Ryan, M.T.; Pfanner, N.; Meisinger, C. Machinery for Protein Sorting and Assembly in the Mitochondrial Outer Membrane. Nature 2003, 424, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Song, E.; Wang, W.; Hsieh, C.-H.; Wang, X.; Feng, W.; Wang, X.; Shen, K. Metaxins Are Core Components of Mitochondrial Transport Adaptor Complexes. Nat. Commun. 2021, 12, 83. [Google Scholar] [CrossRef] [PubMed]
- Abdul, K.M.; Terada, K.; Yano, M.; Ryan, M.T.; Streimann, I.; Hoogenraad, N.J.; Mori, M. Functional Analysis of Human Metaxin in Mitochondrial Protein Import in Cultured Cells and Its Relationship with the Tom Complex. Biochem. Biophys. Res. Commun. 2000, 276, 1028–1034. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, L.C.; Komiya, T.; Bergman, B.E.; Mihara, K.; Bornstein, P. Metaxin Is a Component of a Preprotein Import Complex in the Outer Membrane of the Mammalian Mitochondrion. J. Biol. Chem. 1997, 272, 6510–6518. [Google Scholar] [CrossRef] [PubMed]
- Haider, N.; Dutt, P.; van de Kooij, B.; Ho, J.; Palomero, L.; Pujana, M.A.; Yaffe, M.; Stambolic, V. NEK10 Tyrosine Phosphorylates P53 and Controls Its Transcriptional Activity. Oncogene 2020, 39, 5252–5266. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Fukasawa, Y.; Tsuji, J.; Fu, S.-C.; Tomii, K.; Horton, P.; Imai, K. MitoFates: Improved Prediction of Mitochondrial Targeting Sequences and Their Cleavage Sites. Mol. Cell Proteom. 2015, 14, 1113–1126. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The Multifaceted Contributions of Mitochondria to Cellular Metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Sousa, J.S.; D’Imprima, E.; Vonck, J. Mitochondrial Respiratory Chain Complexes. In Membrane Protein Complexes: Structure and Function; Harris, J.R., Boekema, E.J., Eds.; Subcellular Biochemistry; Springer: Singapore, 2018; Volume 87, pp. 167–227. ISBN 978-981-10-7756-2. [Google Scholar]
- Jonckheere, A.I.; Smeitink, J.A.M.; Rodenburg, R.J.T. Mitochondrial ATP Synthase: Architecture, Function and Pathology. J. Inher Metab. Disea 2012, 35, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Qi, W.; Zou, C.; Xie, Z.; Zhang, M.; Naito, M.G.; Mifflin, L.; Liu, Z.; Najafov, A.; Pan, H.; et al. NEK1-Mediated Retromer Trafficking Promotes Blood–Brain Barrier Integrity by Regulating Glucose Metabolism and RIPK1 Activation. Nat. Commun. 2021, 12, 4826. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Khalil, M.I.; De Benedetti, A. The TLK1/Nek1 Axis Contributes to Mitochondrial Integrity and Apoptosis Prevention via Phosphorylation of VDAC1. Cell Cycle 2020, 19, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Martins, M.B.; Perez, A.M.; Bohr, V.A.; Wilson, D.M., III; Kobarg, J. NEK1 Deficiency Affects Mitochondrial Functions and the Transcriptome of Key DNA Repair Pathways. Mutagenesis 2021, 36, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, W.W.; Chandel, N.S. Hypoxia. 2. Hypoxia Regulates Cellular Metabolism. Am. J. Physiol.-Cell Physiol. 2011, 300, C385–C393. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Zhou, J.; Chen, J.; Zhu, J.; Liu, S.-C.; Ding, X.-F.; Zhang, Q. VHL Regulates NEK1 via Both HIF-2α Pathway and Ubiquitin-Proteasome Pathway in Renal Cancer Cell. Biochem. Biophys. Res. Commun. 2019, 509, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Patil, M.; Pabla, N.; Huang, S.; Dong, Z. Nek1 Phosphorylates Von Hippel-Lindau Tumor Suppressor to Promote Its Proteasomal Degradation and Ciliary Destabilization. Cell Cycle 2013, 12, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Sharma, L.; Lu, J.; Bai, Y. Mitochondrial Respiratory Complex I: Structure, Function and Implication in Human Diseases. CMC 2009, 16, 1266–1277. [Google Scholar] [CrossRef]
- Hadrava Vanova, K.; Kraus, M.; Neuzil, J.; Rohlena, J. Mitochondrial Complex II and Reactive Oxygen Species in Disease and Therapy. Redox Rep. 2020, 25, 26–32. [Google Scholar] [CrossRef]
- Pavan, I.C.B.; Basei, F.L.; Severino, M.B.; Rosa, E.; Silva, I.; Issayama, L.K.; Mancini, M.C.S.; Góis, M.M.; da Silva, L.G.S.; Bezerra, R.M.N.; et al. NEK6 Regulates Redox Balance and DNA Damage Response in DU-145 Prostate Cancer Cells. Cells 2023, 12, 256. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer Metabolism: Fatty Acid Oxidation in the Limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Röhrig, F.; Schulze, A. The Multifaceted Roles of Fatty Acid Synthesis in Cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, G.; Remington, S.J. CITRATE SYNTHASE: Structure, Control, and Mechanism. Annu. Rev. Biophys. Biophys. Chem. 1986, 15, 97–117. [Google Scholar] [CrossRef] [PubMed]
- Alabduladhem, T.O.; Bordoni, B. Physiology, Krebs Cycle. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Srere, P.A. Controls of Citrate Synthase Activity. Life Sci. 1974, 15, 1695–1710. [Google Scholar] [CrossRef]
- Gabriel, B.M.; Al-Tarrah, M.; Alhindi, Y.; Kilikevicius, A.; Venckunas, T.; Gray, S.R.; Lionikas, A.; Ratkevicius, A. H55N Polymorphism Is Associated with Low Citrate Synthase Activity Which Regulates Lipid Metabolism in Mouse Muscle Cells. PLoS ONE 2017, 12, e0185789. [Google Scholar] [CrossRef] [PubMed]
- Alhindi, Y.; Vaanholt, L.M.; Al-Tarrah, M.; Gray, S.R.; Speakman, J.R.; Hambly, C.; Alanazi, B.S.; Gabriel, B.M.; Lionikas, A.; Ratkevicius, A. Low Citrate Synthase Activity Is Associated with Glucose Intolerance and Lipotoxicity. J. Nutr. Metab. 2019, 2019, 8594825. [Google Scholar] [CrossRef] [PubMed]
- Blomme, A.; Ford, C.A.; Mui, E.; Patel, R.; Ntala, C.; Jamieson, L.E.; Planque, M.; McGregor, G.H.; Peixoto, P.; Hervouet, E.; et al. 2,4-Dienoyl-CoA Reductase Regulates Lipid Homeostasis in Treatment-Resistant Prostate Cancer. Nat. Commun. 2020, 11, 2508. [Google Scholar] [CrossRef]
- Nassar, Z.D.; Mah, C.Y.; Dehairs, J.; Burvenich, I.J.; Irani, S.; Centenera, M.M.; Helm, M.; Shrestha, R.K.; Moldovan, M.; Don, A.S.; et al. Human DECR1 Is an Androgen-Repressed Survival Factor That Regulates PUFA Oxidation to Protect Prostate Tumor Cells from Ferroptosis. eLife 2020, 9, e54166. [Google Scholar] [CrossRef]
- Xu, R.; Hu, Q.; Ma, Q.; Liu, C.; Wang, G. The Protease Omi Regulates Mitochondrial Biogenesis through the GSK3β/PGC-1α Pathway. Cell Death Dis. 2014, 5, e1373. [Google Scholar] [CrossRef]
- Yan, J.; Liu, X.-H.; Han, M.-Z.; Wang, Y.-M.; Sun, X.-L.; Yu, N.; Li, T.; Su, B.; Chen, Z.-Y. Blockage of GSK3β-Mediated Drp1 Phosphorylation Provides Neuroprotection in Neuronal and Mouse Models of Alzheimer’s Disease. Neurobiol. Aging 2015, 36, 211–227. [Google Scholar] [CrossRef] [PubMed]
- Papadopoli, D.; Pollak, M.; Topisirovic, I. The Role of GSK3 in Metabolic Pathway Perturbations in Cancer. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2021, 1868, 119059. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.Q.; Li, C.; Stanley, C.A.; Smith, T.J. Glutamate Dehydrogenase, a Complex Enzyme at a Crucial Metabolic Branch Point. Neurochem. Res. 2019, 44, 117–132. [Google Scholar] [CrossRef]
- Luo, M.; Ma, W.; Zapata-Bustos, R.; Willis, W.T.; Mandarino, L.J. Deletion of Von Willebrand A Domain Containing Protein (VWA8) Raises Activity of Mitochondrial Electron Transport Chain Complexes in Hepatocytes. Biochem. Biophys. Rep. 2021, 26, 100928. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, J.P.; Basei, F.L.; Rojas, M.L.; Galvis, D.; Zorzano, A. Mechanisms of Modulation of Mitochondrial Architecture. Biomolecules 2023, 13, 1225. [Google Scholar] [CrossRef] [PubMed]
- Giacomello, M.; Pyakurel, A.; Glytsou, C.; Scorrano, L. The Cell Biology of Mitochondrial Membrane Dynamics. Nat. Rev. Mol. Cell Biol. 2020, 21, 204–224. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wang, L.; Yang, C.; Pu, S.; Guo, Z.; Wu, Q.; Zhou, Z.; Zhao, H. Mitochondrial Membrane Remodeling. Front. Bioeng. Biotechnol. 2022, 9, 786806. [Google Scholar] [CrossRef]
- Twig, G.; Elorza, A.; Molina, A.J.A.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and Selective Fusion Govern Mitochondrial Segregation and Elimination by Autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic Phosphorylation of Dynamin-Related GTPase Drp1 Participates in Mitochondrial Fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef]
- Kanfer, G.; Kornmann, B. Dynamics of the Mitochondrial Network during Mitosis. Biochem. Soc. Trans. 2016, 44, 510–516. [Google Scholar] [CrossRef]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff Is an Essential Factor for Mitochondrial Recruitment of Drp1 during Mitochondrial Fission in Mammalian Cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Cipolat, S.; de Brito, O.M.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; Strooper, B.D.; et al. OPA1 Controls Apoptotic Cristae Remodeling Independently from Mitochondrial Fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Patten, D.A.; Wong, J.; Khacho, M.; Soubannier, V.; Mailloux, R.J.; Pilon-Larose, K.; MacLaurin, J.G.; Park, D.S.; McBride, H.M.; Trinkle-Mulcahy, L.; et al. OPA1-dependent Cristae Modulation Is Essential for Cellular Adaptation to Metabolic Demand. EMBO J. 2014, 33, 2676–2691. [Google Scholar] [CrossRef] [PubMed]
- Holthuis, J.C.M.; Menon, A.K. Lipid Landscapes and Pipelines in Membrane Homeostasis. Nature 2014, 510, 48–57. [Google Scholar] [CrossRef]
- SLAGEN, Consortium; Kenna, K.P.; van Doormaal, P.T.C.; Dekker, A.M.; Ticozzi, N.; Kenna, B.J.; Diekstra, F.P.; van Rheenen, W.; van Eijk, K.R.; Jones, A.R.; et al. NEK1 Variants Confer Susceptibility to Amyotrophic Lateral Sclerosis. Nat. Genet. 2016, 48, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.; Müller, K.; Wieland, T.; Weydt, P.; Böhm, S.; Lulé, D.; Hübers, A.; Neuwirth, C.; Weber, M.; Borck, G.; et al. NEK1 Mutations in Familial Amyotrophic Lateral Sclerosis. Brain 2016, 139, e28. [Google Scholar] [CrossRef] [PubMed]
- Benson, B.C.; Shaw, P.J.; Azzouz, M.; Highley, J.R.; Hautbergue, G.M. Proteinopathies as Hallmarks of Impaired Gene Expression, Proteostasis and Mitochondrial Function in Amyotrophic Lateral Sclerosis. Front. Neurosci. 2021, 15, 783624. [Google Scholar] [CrossRef]
- Dupuis, L.; Gonzalez de Aguilar, J.-L.; Oudart, H.; de Tapia, M.; Barbeito, L.; Loeffler, J.-P. Mitochondria in Amyotrophic Lateral Sclerosis: A Trigger and a Target. Neurodegener. Dis. 2004, 1, 245–254. [Google Scholar] [CrossRef]
- Amiri, M.; Hollenbeck, P.J. Mitochondrial Biogenesis in the Axons of Vertebrate Peripheral Neurons. Dev. Neurobiol. 2008, 68, 1348–1361. [Google Scholar] [CrossRef]
- Brodin, L.; Shupliakov, O. Retromer in Synaptic Function and Pathology. Front. Synaptic Neurosci. 2018, 10, 37. [Google Scholar] [CrossRef]
- Liesa, M.; Shirihai, O.S. Mitochondrial Dynamics in the Regulation of Nutrient Utilization and Energy Expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef] [PubMed]
- Molina, A.J.A.; Wikstrom, J.D.; Stiles, L.; Las, G.; Mohamed, H.; Elorza, A.; Walzer, G.; Twig, G.; Katz, S.; Corkey, B.E.; et al. Mitochondrial Networking Protects β-Cells From Nutrient-Induced Apoptosis. Diabetes 2009, 58, 2303–2315. [Google Scholar] [CrossRef]
- Wikstrom, J.D.; Mahdaviani, K.; Liesa, M.; Sereda, S.B.; Si, Y.; Las, G.; Twig, G.; Petrovic, N.; Zingaretti, C.; Graham, A.; et al. Hormone-Induced Mitochondrial Fission Is Utilized by Brown Adipocytes as an Amplification Pathway for Energy Expenditure. EMBO J. 2014, 33, 418–436. [Google Scholar] [CrossRef] [PubMed]
- Shields, L.Y.; Kim, H.; Zhu, L.; Haddad, D.; Berthet, A.; Pathak, D.; Lam, M.; Ponnusamy, R.; Diaz-Ramirez, L.G.; Gill, T.M.; et al. Dynamin-Related Protein 1 Is Required for Normal Mitochondrial Bioenergetic and Synaptic Function in CA1 Hippocampal Neurons. Cell Death Dis. 2015, 6, e1725. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, N.; Nomura, M.; Jofuku, A.; Kato, H.; Suzuki, S.O.; Masuda, K.; Otera, H.; Nakanishi, Y.; Nonaka, I.; Goto, Y.-I.; et al. Mitochondrial Fission Factor Drp1 Is Essential for Embryonic Development and Synapse Formation in Mice. Nat. Cell Biol. 2009, 11, 958–966. [Google Scholar] [CrossRef]
- Nan, J.; Lee, J.S.; Moon, J.H.; Lee, S.-A.; Park, Y.J.; Lee, D.-S.; Chung, S.S.; Park, K.S. SENP2 Regulates Mitochondrial Function and Insulin Secretion in Pancreatic β Cells. Exp. Mol. Med. 2022, 54, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Kleele, T.; Rey, T.; Winter, J.; Zaganelli, S.; Mahecic, D.; Perreten Lambert, H.; Ruberto, F.P.; Nemir, M.; Wai, T.; Pedrazzini, T.; et al. Distinct Fission Signatures Predict Mitochondrial Degradation or Biogenesis. Nature 2021, 593, 435–439. [Google Scholar] [CrossRef]
- Chang, Y.-J.; Chen, K.-W.; Chen, L. Mitochondrial ROS1 Increases Mitochondrial Fission and Respiration in Oral Squamous Cancer Carcinoma. Cancers 2020, 12, 2845. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, L.; Jia, R. The Role of Mitochondrial Dynamics in Human Cancers. Am. J. Cancer Res. 2020, 10, 1278–1293. [Google Scholar]
- He, B.; Yu, H.; Liu, S.; Wan, H.; Fu, S.; Liu, S.; Yang, J.; Zhang, Z.; Huang, H.; Li, Q.; et al. Mitochondrial Cristae Architecture Protects against mtDNA Release and Inflammation. Cell Rep. 2022, 41, 111774. [Google Scholar] [CrossRef]
- Li, S.; Rousseau, D. ATAD3, a Vital Membrane Bound Mitochondrial ATPase Involved in Tumor Progression. J. Bioenerg. Biomembr. 2012, 44, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Peralta, S.; Goffart, S.; Williams, S.L.; Diaz, F.; Garcia, S.; Nissanka, N.; Area-Gomez, E.; Pohjoismäki, J.; Moraes, C.T. ATAD3 Controls Mitochondrial Cristae Structure in Mouse Muscle, Influencing mtDNA Replication and Cholesterol Levels. J. Cell Sci. 2018, 131, jcs217075. [Google Scholar] [CrossRef]
- Paumard, P.; Vaillier, J.; Coulary, B.; Schaeffer, J.; Soubannier, V.; Mueller, D.M.; Brèthes, D.; di Rago, J.-P.; Velours, J. The ATP Synthase Is Involved in Generating Mitochondrial Cristae Morphology. EMBO J. 2002, 21, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Baudier, J. ATAD3 Proteins: Brokers of a Mitochondria-Endoplasmic Reticulum Connection in Mammalian Cells: Mitochondria-Endoplasmic Reticulum Connection. Biol. Rev. 2018, 93, 827–844. [Google Scholar] [CrossRef] [PubMed]
- Richter-Dennerlein, R.; Korwitz, A.; Haag, M.; Tatsuta, T.; Dargazanli, S.; Baker, M.; Decker, T.; Lamkemeyer, T.; Rugarli, E.I.; Langer, T. DNAJC19, a Mitochondrial Cochaperone Associated with Cardiomyopathy, Forms a Complex with Prohibitins to Regulate Cardiolipin Remodeling. Cell Metab. 2014, 20, 158–171. [Google Scholar] [CrossRef]
- Machiraju, P.; Wang, X.; Sabouny, R.; Huang, J.; Zhao, T.; Iqbal, F.; King, M.; Prasher, D.; Lodha, A.; Jimenez-Tellez, N.; et al. SS-31 Peptide Reverses the Mitochondrial Fragmentation Present in Fibroblasts From Patients With DCMA, a Mitochondrial Cardiomyopathy. Front. Cardiovasc. Med. 2019, 6, 167. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and Organization of the Human Mitochondrial Genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- Garrido, N.; Griparic, L.; Jokitalo, E.; Wartiovaara, J.; van der Bliek, A.M.; Spelbrink, J.N. Composition and Dynamics of Human Mitochondrial Nucleoids. MBoC 2003, 14, 1583–1596. [Google Scholar] [CrossRef]
- Legros, F.; Malka, F.; Frachon, P.; Lombès, A.; Rojo, M. Organization and Dynamics of Human Mitochondrial DNA. J. Cell Sci. 2004, 117, 2653–2662. [Google Scholar] [CrossRef]
- Korhonen, J.A.; Pham, X.H.; Pellegrini, M.; Falkenberg, M. Reconstitution of a Minimal mtDNA Replisome in Vitro. EMBO J. 2004, 23, 2423–2429. [Google Scholar] [CrossRef]
- Richter, C.; Park, J.W.; Ames, B.N. Normal Oxidative Damage to Mitochondrial and Nuclear DNA Is Extensive. Proc. Natl. Acad. Sci. USA 1988, 85, 6465–6467. [Google Scholar] [CrossRef] [PubMed]
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA Damage Is More Extensive and Persists Longer than Nuclear DNA Damage in Human Cells Following Oxidative Stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Alencar, R.R.; Batalha, C.M.P.F.; Freire, T.S.; de Souza-Pinto, N.C. Enzymology of Mitochondrial DNA Repair. Enzymes 2019, 45, 257–287. [Google Scholar] [CrossRef]
- Rashid, S.; Freitas, M.O.; Cucchi, D.; Bridge, G.; Yao, Z.; Gay, L.; Williams, M.; Wang, J.; Suraweera, N.; Silver, A.; et al. MLH1 Deficiency Leads to Deregulated Mitochondrial Metabolism. Cell Death Dis. 2019, 10, 795. [Google Scholar] [CrossRef] [PubMed]
- de Souza-Pinto, N.C.; Mason, P.A.; Hashiguchi, K.; Weissman, L.; Tian, J.; Guay, D.; Lebel, M.; Stevnsner, T.V.; Rasmussen, L.J.; Bohr, V.A. Novel DNA Mismatch-Repair Activity Involving YB-1 in Human Mitochondria. DNA Repair. 2009, 8, 704–719. [Google Scholar] [CrossRef] [PubMed]
- Cleaver, J.E.; Brennan-Minnella, A.M.; Swanson, R.A.; Fong, K.; Chen, J.; Chou, K.; Chen, Y.; Revet, I.; Bezrookove, V. Mitochondrial Reactive Oxygen Species Are Scavenged by Cockayne Syndrome B Protein in Human Fibroblasts without Nuclear DNA Damage. Proc. Natl. Acad. Sci. USA 2014, 111, 13487–13492. [Google Scholar] [CrossRef] [PubMed]
- Aamann, M.D.; Sorensen, M.M.; Hvitby, C.; Berquist, B.R.; Muftuoglu, M.; Tian, J.; Souza-Pinto, N.C.; Scheibye-Knudsen, M.; Wilson, D.M.; Stevnsner, T.; et al. Cockayne Syndrome Group B Protein Promotes Mitochondrial DNA Stability by Supporting the DNA Repair Association with the Mitochondrial Membrane. FASEB J. 2010, 24, 2334–2346. [Google Scholar] [CrossRef]
- Moretton, A.; Morel, F.; Macao, B.; Lachaume, P.; Ishak, L.; Lefebvre, M.; Garreau-Balandier, I.; Vernet, P.; Falkenberg, M.; Farge, G. Selective Mitochondrial DNA Degradation Following Double-Strand Breaks. PLoS ONE 2017, 12, e0176795. [Google Scholar] [CrossRef]
- Coffey, G. An Alternate Form of Ku80 Is Required for DNA End-Binding Activity in Mammalian Mitochondria. Nucleic Acids Res. 2000, 28, 3793–3800. [Google Scholar] [CrossRef]
- Tadi, S.K.; Sebastian, R.; Dahal, S.; Babu, R.K.; Choudhary, B.; Raghavan, S.C. Microhomology-Mediated End Joining Is the Principal Mediator of Double-Strand Break Repair during Mitochondrial DNA Lesions. Mol. Biol. Cell 2016, 27, 223–235. [Google Scholar] [CrossRef]
- Dahal, S.; Dubey, S.; Raghavan, S.C. Homologous Recombination-Mediated Repair of DNA Double-Strand Breaks Operates in Mammalian Mitochondria. Cell. Mol. Life Sci. 2018, 75, 1641–1655. [Google Scholar] [CrossRef]
- Gredilla, R.; Garm, C.; Stevnsner, T. Nuclear and Mitochondrial DNA Repair in Selected Eukaryotic Aging Model Systems. Oxid. Med. Cell. Longev. 2012, 2012, 282438. [Google Scholar] [CrossRef]
- Yasukawa, T.; Kang, D. An Overview of Mammalian Mitochondrial DNA Replication Mechanisms. J. Biochem. 2018, 164, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Ekstrand, M.I. Mitochondrial Transcription Factor A Regulates mtDNA Copy Number in Mammals. Human. Mol. Genet. 2004, 13, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Kanki, T.; Ohgaki, K.; Gaspari, M.; Gustafsson, C.M.; Fukuoh, A.; Sasaki, N.; Hamasaki, N.; Kang, D. Architectural Role of Mitochondrial Transcription Factor A in Maintenance of Human Mitochondrial DNA. Mol. Cell. Biol. 2004, 24, 9823–9834. [Google Scholar] [CrossRef]
- Filograna, R.; Mennuni, M.; Alsina, D.; Larsson, N. Mitochondrial DNA Copy Number in Human Disease: The More the Better? FEBS Lett. 2021, 595, 976–1002. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Xie, X.; Wang, Y.; Gao, Y.; Xie, X.; Yang, J.; Ye, J. Association between Leukocyte Mitochondrial DNA Content and Risk of Coronary Heart Disease: A Case-Control Study. Atherosclerosis 2014, 237, 220–226. [Google Scholar] [CrossRef]
- Tin, A.; Grams, M.E.; Ashar, F.N.; Lane, J.A.; Rosenberg, A.Z.; Grove, M.L.; Boerwinkle, E.; Selvin, E.; Coresh, J.; Pankratz, N.; et al. Association between Mitochondrial DNA Copy Number in Peripheral Blood and Incident CKD in the Atherosclerosis Risk in Communities Study. J. Am. Soc. Nephrol. 2016, 27, 2467. [Google Scholar] [CrossRef]
- Zhang, Y.; Guallar, E.; Ashar, F.N.; Longchamps, R.J.; Castellani, C.A.; Lane, J.; Grove, M.L.; Coresh, J.; Sotoodehnia, N.; Ilkhanoff, L.; et al. Association between Mitochondrial DNA Copy Number and Sudden Cardiac Death: Findings from the Atherosclerosis Risk in Communities Study (ARIC). Eur. Heart J. 2017, 38, 3443–3448. [Google Scholar] [CrossRef]
- Polci, R.; Peng, A.; Chen, P.-L.; Riley, D.J.; Chen, Y. NIMA-Related Protein Kinase 1 Is Involved Early in the Ionizing Radiation-Induced DNA Damage Response. Cancer Res. 2004, 64, 8800–8803. [Google Scholar] [CrossRef]
- Liu, S.; Ho, C.K.; Ouyang, J.; Zou, L. Nek1 Kinase Associates with ATR-ATRIP and Primes ATR for Efficient DNA Damage Signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 2175–2180. [Google Scholar] [CrossRef]
- Nguyen, C.L.; Possemato, R.; Bauerlein, E.L.; Xie, A.; Scully, R.; Hahn, W.C. Nek4 Regulates Entry into Replicative Senescence and the Response to DNA Damage in Human Fibroblasts. Mol. Cell. Biol. 2012, 32, 3963–3977. [Google Scholar] [CrossRef] [PubMed]
- Melo-Hanchuk, T.D.; Slepicka, P.F.; Pelegrini, A.L.; Menck, C.F.M.; Kobarg, J. NEK5 Interacts with Topoisomerase IIβ and Is Involved in the DNA Damage Response Induced by Etoposide. J. Cell. Biochem. 2019, 120, 16853–16866. [Google Scholar] [CrossRef] [PubMed]
- Sobek, S.; Boege, F. DNA Topoisomerases in mtDNA Maintenance and Ageing. Exp. Gerontol. 2014, 56, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Lee, J.; Nie, X.; Li, M.; Morozov, Y.I.; Venkatesh, S.; Bogenhagen, D.F.; Temiakov, D.; Suzuki, C.K. Phosphorylation of Human TFAM in Mitochondria Impairs DNA Binding and Promotes Degradation by the AAA+ Lon Protease. Mol. Cell 2013, 49, 121–132. [Google Scholar] [CrossRef] [PubMed]
- de Souza-Pinto, N.C.; Wilson, D.M.; Stevnsner, T.V.; Bohr, V.A. Mitochondrial DNA, Base Excision Repair and Neurodegeneration. DNA Repair. 2008, 7, 1098–1109. [Google Scholar] [CrossRef] [PubMed]
- Canugovi, C.; Maynard, S.; Bayne, A.-C.V.; Sykora, P.; Tian, J.; de Souza-Pinto, N.C.; Croteau, D.L.; Bohr, V.A. The Mitochondrial Transcription Factor A Functions in Mitochondrial Base Excision Repair. DNA Repair. 2010, 9, 1080–1089. [Google Scholar] [CrossRef] [PubMed]
- Bazzani, V.; Barchiesi, A.; Radecka, D.; Pravisani, R.; Guadagno, A.; Di Loreto, C.; Baccarani, U.; Vascotto, C. Mitochondrial Apurinic/Apyrimidinic Endonuclease 1 Enhances mtDNA Repair Contributing to Cell Proliferation and Mitochondrial Integrity in Early Stages of Hepatocellular Carcinoma. BMC Cancer 2020, 20, 969. [Google Scholar] [CrossRef]
- Matsushima, Y.; Goto, Y.; Kaguni, L.S. Mitochondrial Lon Protease Regulates Mitochondrial DNA Copy Number and Transcription by Selective Degradation of Mitochondrial Transcription Factor A (TFAM). Proc. Natl. Acad. Sci. USA 2010, 107, 18410–18415. [Google Scholar] [CrossRef]
- Matsushima, Y.; Kaguni, L.S. Matrix Proteases in Mitochondrial DNA Function. Biochim. Biophys. Acta (BBA)—Gene Regul. Mech. 2012, 1819, 1080–1087. [Google Scholar] [CrossRef]
- Litonin, D.; Sologub, M.; Shi, Y.; Savkina, M.; Anikin, M.; Falkenberg, M.; Gustafsson, C.M.; Temiakov, D. Human Mitochondrial Transcription Revisited. J. Biol. Chem. 2010, 285, 18129–18133. [Google Scholar] [CrossRef]
- Kondadi, A.K.; Anand, R.; Reichert, A.S. Functional Interplay between Cristae Biogenesis, Mitochondrial Dynamics and Mitochondrial DNA Integrity. Int. J. Mol. Sci. 2019, 20, 4311. [Google Scholar] [CrossRef]
- Li, H.; Ruan, Y.; Zhang, K.; Jian, F.; Hu, C.; Miao, L.; Gong, L.; Sun, L.; Zhang, X.; Chen, S.; et al. Mic60/Mitofilin Determines MICOS Assembly Essential for Mitochondrial Dynamics and mtDNA Nucleoid Organization. Cell Death Differ. 2016, 23, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Lepelley, A.; Della Mina, E.; Van Nieuwenhove, E.; Waumans, L.; Fraitag, S.; Rice, G.I.; Dhir, A.; Frémond, M.-L.; Rodero, M.P.; Seabra, L.; et al. Enhanced cGAS-STING–Dependent Interferon Signaling Associated with Mutations in ATAD3A. J. Exp. Med. 2021, 218, e20201560. [Google Scholar] [CrossRef] [PubMed]
- Ashton, N.W.; Bolderson, E.; Cubeddu, L.; O’Byrne, K.J.; Richard, D.J. Human Single-Stranded DNA Binding Proteins Are Essential for Maintaining Genomic Stability. BMC Mol. Biol. 2013, 14, 9. [Google Scholar] [CrossRef] [PubMed]
- Rong, Z.; Tu, P.; Xu, P.; Sun, Y.; Yu, F.; Tu, N.; Guo, L.; Yang, Y. The Mitochondrial Response to DNA Damage. Front. Cell Dev. Biol. 2021, 9, 669379. [Google Scholar] [CrossRef] [PubMed]
- Caston, R.A.; Gampala, S.; Armstrong, L.; Messmann, R.A.; Fishel, M.L.; Kelley, M.R. The Multifunctional APE1 DNA Repair–Redox Signaling Protein as a Drug Target in Human Disease. Drug Discov. Today 2021, 26, 218–228. [Google Scholar] [CrossRef]
- Whitaker, A.M.; Freudenthal, B.D. APE1: A Skilled Nucleic Acid Surgeon. DNA Repair. 2018, 71, 93–100. [Google Scholar] [CrossRef]
- Chen, W.; Wang, S.; Xing, D. New Horizons for the Roles and Association of APE1/Ref-1 and ABCA1 in Atherosclerosis. J. Inflamm. Res. 2021, 14, 5251–5271. [Google Scholar] [CrossRef]
- Kunová, N.; Ondrovičová, G.; Bauer, J.A.; Bellová, J.; Ambro, Ľ.; Martináková, L.; Kotrasová, V.; Kutejová, E.; Pevala, V. The Role of Lon-Mediated Proteolysis in the Dynamics of Mitochondrial Nucleic Acid-Protein Complexes. Sci. Rep. 2017, 7, 631. [Google Scholar] [CrossRef]
- Bedoui, S.; Herold, M.J.; Strasser, A. Emerging Connectivity of Programmed Cell Death Pathways and Its Physiological Implications. Nat. Rev. Mol. Cell Biol. 2020, 21, 678–695. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.K.; Sethi, G. Molecular Mechanisms of Cell Death. In Mechanisms of Cell Death and Opportunities for Therapeutic Development; Elsevier: Amsterdam, The Netherlands, 2022; pp. 65–92. ISBN 978-0-12-814208-0. [Google Scholar]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The Molecular Machinery of Regulated Cell Death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Bock, F.J.; Tait, S.W.G. Mitochondria as Multifaceted Regulators of Cell Death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Su, S.S.; Zhao, S.; Yang, Z.; Zhong, C.-Q.; Chen, X.; Cai, Q.; Yang, Z.-H.; Huang, D.; Wu, R.; et al. RIP1 Autophosphorylation Is Promoted by Mitochondrial ROS and Is Essential for RIP3 Recruitment into Necrosome. Nat. Commun. 2017, 8, 14329. [Google Scholar] [CrossRef] [PubMed]
- Bhola, P.D.; Mattheyses, A.L.; Simon, S.M. Spatial and Temporal Dynamics of Mitochondrial Membrane Permeability Waves during Apoptosis. Biophys. J. 2009, 97, 2222–2231. [Google Scholar] [CrossRef]
- Goldstein, J.C.; Waterhouse, N.J.; Juin, P.; Evan, G.I.; Green, D.R. The Coordinate Release of Cytochrome c during Apoptosis Is Rapid, Complete and Kinetically Invariant. Nat. Cell Biol. 2000, 2, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Colell, A.; Ricci, J.-E.; Tait, S.; Milasta, S.; Maurer, U.; Bouchier-Hayes, L.; Fitzgerald, P.; Guio-Carrion, A.; Waterhouse, N.J.; Li, C.W.; et al. GAPDH and Autophagy Preserve Survival after Apoptotic Cytochrome c Release in the Absence of Caspase Activation. Cell 2007, 129, 983–997. [Google Scholar] [CrossRef]
- Seo, J.H.; Chae, Y.C.; Kossenkov, A.V.; Lee, Y.G.; Tang, H.-Y.; Agarwal, E.; Gabrilovich, D.I.; Languino, L.R.; Speicher, D.W.; Shastrula, P.K.; et al. MFF Regulation of Mitochondrial Cell Death Is a Therapeutic Target in Cancer. Cancer Res. 2019, 79, 6215–6226. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, C.-F.; Polci, R.; Wei, R.; Riley, D.J.; Chen, P.-L. Increased Nek1 Expression in Renal Cell Carcinoma Cells Is Associated with Decreased Sensitivity to DNA-Damaging Treatment. Oncotarget 2014, 5, 4283–4294. [Google Scholar] [CrossRef]
- Pelegrini, A.L.; Moura, D.J.; Brenner, B.L.; Ledur, P.F.; Maques, G.P.; Henriques, J.A.P.; Saffi, J.; Lenz, G. Nek1 Silencing Slows down DNA Repair and Blocks DNA Damage-Induced Cell Cycle Arrest. Mutagenesis 2010, 25, 447–454. [Google Scholar] [CrossRef]
- Schenk, B.; Fulda, S. Reactive Oxygen Species Regulate Smac Mimetic/TNFα-Induced Necroptotic Signaling and Cell Death. Oncogene 2015, 34, 5796–5806. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Sawasaki, T. Nek5, a Novel Substrate for Caspase-3, Promotes Skeletal Muscle Differentiation by up-Regulating Caspase Activity. FEBS Lett. 2013, 587, 2219–2225. [Google Scholar] [CrossRef]
- Park, S.J.; Jo, D.S.; Jo, S.-Y.; Shin, D.W.; Shim, S.; Jo, Y.K.; Shin, J.H.; Ha, Y.J.; Jeong, S.-Y.; Hwang, J.J.; et al. Inhibition of Never in Mitosis A (NIMA)-Related Kinase-4 Reduces Survivin Expression and Sensitizes Cancer Cells to TRAIL-Induced Cell Death. Oncotarget 2016, 7, 65957–65967. [Google Scholar] [CrossRef] [PubMed]
- Doles, J.; Hemann, M.T. Nek4 Status Differentially Alters Sensitivity to Distinct Microtubule Poisons. Cancer Res. 2010, 70, 1033–1041. [Google Scholar] [CrossRef] [PubMed]
- Jastroch, M.; Divakaruni, A.S.; Mookerjee, S.; Treberg, J.R.; Brand, M.D. Mitochondrial Proton and Electron Leaks. Essays Biochem. 2010, 47, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Matoba, S.; Kang, J.-G.; Patino, W.D.; Wragg, A.; Boehm, M.; Gavrilova, O.; Hurley, P.J.; Bunz, F.; Hwang, P.M. P53 Regulates Mitochondrial Respiration. Science 2006, 312, 1650–1653. [Google Scholar] [CrossRef]
- Daugas, E.; Susin, S.A.; Zamzami, N.; Ferri, K.F.; Irinopoulou, T.; Larochette, N.; Prévost, M.C.; Leber, B.; Andrews, D.; Penninger, J.; et al. Mitochondrio-Nuclear Translocation of AIF in Apoptosis and Necrosis. FASEB J. 2000, 14, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.-W.; Wang, H.; Poitras, M.F.; Coombs, C.; Bowers, W.J.; Federoff, H.J.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Mediation of Poly(ADP-Ribose) Polymerase-1-Dependent Cell Death by Apoptosis-Inducing Factor. Science 2002, 297, 259–263. [Google Scholar] [CrossRef]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the Integrated Stress Response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef]
- He, C.; Klionsky, D.J. Regulation Mechanisms and Signaling Pathways of Autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef]
- Muñoz, J.P.; Ivanova, S.; Sánchez-Wandelmer, J.; Martínez-Cristóbal, P.; Noguera, E.; Sancho, A.; Díaz-Ramos, A.; Hernández-Alvarez, M.I.; Sebastián, D.; Mauvezin, C.; et al. Mfn2 Modulates the UPR and Mitochondrial Function via Repression of PERK. EMBO J. 2013, 32, 2348–2361. [Google Scholar] [CrossRef] [PubMed]
- Rainbolt, T.K.; Saunders, J.M.; Wiseman, R.L. Stress-Responsive Regulation of Mitochondria through the ER Unfolded Protein Response. Trends Endocrinol. Metab. 2014, 25, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of Mammalian Autophagy in Physiology and Pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [PubMed]
- Tang, A.H.; Rando, T.A. Induction of Autophagy Supports the Bioenergetic Demands of Quiescent Muscle Stem Cell Activation. EMBO J. 2014, 33, 2782–2797. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. Eaten Alive: A History of Macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Elazar, Z. Mechanism and Medical Implications of Mammalian Autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of Mitophagy in Cellular Homeostasis, Physiology and Pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 Conjugation System in Mammalian Autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2503–2518. [Google Scholar] [CrossRef]
- Martinus, R.D.; Garth, G.P.; Webster, T.L.; Cartwright, P.; Naylor, D.J.; Høj, P.B.; Hoogenraad, N.J. Selective Induction of Mitochondrial Chaperones in Response to Loss of the Mitochondrial Genome. Eur. J. Biochem. 1996, 240, 98–103. [Google Scholar] [CrossRef]
- Zhao, Q.; Wang, J.; Levichkin, I.V.; Stasinopoulos, S.; Ryan, M.T.; Hoogenraad, N.J. A Mitochondrial Specific Stress Response in Mammalian Cells. EMBO J. 2002, 21, 4411–4419. [Google Scholar] [CrossRef]
- Zhu, L.; Zhou, Q.; He, L.; Chen, L. Mitochondrial Unfolded Protein Response: An Emerging Pathway in Human Diseases. Free Radic. Biol. Med. 2021, 163, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Melber, A.; Haynes, C.M. UPRmt Regulation and Output: A Stress Response Mediated by Mitochondrial-Nuclear Communication. Cell Res. 2018, 28, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Senft, D.; Ronai, Z.A. UPR, Autophagy, and Mitochondria Crosstalk Underlies the ER Stress Response. Trends Biochem. Sci. 2015, 40, 141–148. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Chino, H.; Tsukamoto, S.; Ode, K.L.; Ueda, H.R.; Mizushima, N. NEK9 Regulates Primary Cilia Formation by Acting as a Selective Autophagy Adaptor for MYH9/Myosin IIA. Nat. Commun. 2021, 12, 3292. [Google Scholar] [CrossRef] [PubMed]
- Morleo, M.; Franco, B. The Autophagy-Cilia Axis: An Intricate Relationship. Cells 2019, 8, 905. [Google Scholar] [CrossRef] [PubMed]
- Pampliega, O.; Cuervo, A.M. Autophagy and Primary Cilia: Dual Interplay. Curr. Opin. Cell Biol. 2016, 39, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, B.K.; Skytte Rasmussen, M.; Abudu, Y.P.; Bruun, J.-A.; Larsen, K.B.; Alemu, E.A.; Sjøttem, E.; Lamark, T.; Johansen, T. NIMA-Related Kinase 9–Mediated Phosphorylation of the Microtubule-Associated LC3B Protein at Thr-50 Suppresses Selective Autophagy of P62/Sequestosome 1. J. Biol. Chem. 2020, 295, 1240–1260. [Google Scholar] [CrossRef]
- Xia, J.; He, Y.; Meng, B.; Chen, S.; Zhang, J.; Wu, X.; Zhu, Y.; Shen, Y.; Feng, X.; Guan, Y.; et al. NEK2 Induces Autophagy-mediated Bortezomib Resistance by Stabilizing Beclin-1 in Multiple Myeloma. Mol. Oncol. 2020, 14, 763–778. [Google Scholar] [CrossRef]
- Szyniarowski, P.; Corcelle-Termeau, E.; Farkas, T.; Høyer-Hansen, M.; Nylandsted, J.; Kallunki, T.; Jäättelä, M. A Comprehensive siRNA Screen for Kinases That Suppress Macroautophagy in Optimal Growth Conditions. Autophagy 2011, 7, 892–903. [Google Scholar] [CrossRef]
- Madureira, M.; Connor-Robson, N.; Wade-Martins, R. LRRK2: Autophagy and Lysosomal Activity. Front. Neurosci. 2020, 14, 498. [Google Scholar] [CrossRef]
- Velikkakath, A.K.G.; Nishimura, T.; Oita, E.; Ishihara, N.; Mizushima, N. Mammalian Atg2 Proteins Are Essential for Autophagosome Formation and Important for Regulation of Size and Distribution of Lipid Droplets. MBoC 2012, 23, 896–909. [Google Scholar] [CrossRef] [PubMed]
- N’Diaye, E.; Kajihara, K.K.; Hsieh, I.; Morisaki, H.; Debnath, J.; Brown, E.J. PLIC Proteins or Ubiquilins Regulate Autophagy-dependent Cell Survival during Nutrient Starvation. EMBO Rep. 2009, 10, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.J.; Cai, A.; Greenslade, J.E.; Higgins, N.R.; Fan, C.; Le, N.T.T.; Tatman, M.; Whiteley, A.M.; Prado, M.A.; Dieriks, B.V.; et al. ALS/FTD Mutations in UBQLN2 Impede Autophagy by Reducing Autophagosome Acidification through Loss of Function. Proc. Natl. Acad. Sci. USA 2020, 117, 15230–15241. [Google Scholar] [CrossRef] [PubMed]
- Şentürk, M.; Lin, G.; Zuo, Z.; Mao, D.; Watson, E.; Mikos, A.G.; Bellen, H.J. Ubiquilins Regulate Autophagic Flux through mTOR Signalling and Lysosomal Acidification. Nat. Cell Biol. 2019, 21, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; He, H.; Tang, Z.; Hattori, T.; Liu, Y.; Young, M.M.; Serfass, J.M.; Chen, L.; Gebru, M.; Chen, C.; et al. An Autophagy Assay Reveals the ESCRT-III Component CHMP2A as a Regulator of Phagophore Closure. Nat. Commun. 2018, 9, 2855. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.H. TRAP1 Regulation of Mitochondrial Life or Death Decision in Cancer Cells and Mitochondria-Targeted TRAP1 Inhibitors. BMB Rep. 2012, 45, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Amoroso, M.R.; Matassa, D.S.; Laudiero, G.; Egorova, A.V.; Polishchuk, R.S.; Maddalena, F.; Piscazzi, A.; Paladino, S.; Sarnataro, D.; Garbi, C.; et al. TRAP1 and the Proteasome Regulatory Particle TBP7/Rpt3 Interact in the Endoplasmic Reticulum and Control Cellular Ubiquitination of Specific Mitochondrial Proteins. Cell Death Differ. 2012, 19, 592–604. [Google Scholar] [CrossRef]
- Takamura, H.; Koyama, Y.; Matsuzaki, S.; Yamada, K.; Hattori, T.; Miyata, S.; Takemoto, K.; Tohyama, M.; Katayama, T. TRAP1 Controls Mitochondrial Fusion/Fission Balance through Drp1 and Mff Expression. PLoS ONE 2012, 7, e51912. [Google Scholar] [CrossRef]
- Vainberg, I.E.; Lewis, S.A.; Rommelaere, H.; Ampe, C.; Vandekerckhove, J.; Klein, H.L.; Cowan, N.J. Prefoldin, a Chaperone That Delivers Unfolded Proteins to Cytosolic Chaperonin. Cell 1998, 93, 863–873. [Google Scholar] [CrossRef]
- Takano, M.; Tashiro, E.; Kitamura, A.; Maita, H.; Iguchi-Ariga, S.M.M.; Kinjo, M.; Ariga, H. Prefoldin Prevents Aggregation of α-Synuclein. Brain Res. 2014, 1542, 186–194. [Google Scholar] [CrossRef]
- Tashiro, E.; Zako, T.; Muto, H.; Itoo, Y.; Sörgjerd, K.; Terada, N.; Abe, A.; Miyazawa, M.; Kitamura, A.; Kitaura, H.; et al. Prefoldin Protects Neuronal Cells from Polyglutamine Toxicity by Preventing Aggregation Formation. J. Biol. Chem. 2013, 288, 19958–19972. [Google Scholar] [CrossRef]
- Lovett, D.H.; Mahimkar, R.; Raffai, R.L.; Cape, L.; Maklashina, E.; Cecchini, G.; Karliner, J.S. A Novel Intracellular Isoform of Matrix Metalloproteinase-2 Induced by Oxidative Stress Activates Innate Immunity. PLoS ONE 2012, 7, e34177. [Google Scholar] [CrossRef] [PubMed]
- Pangou, E.; Sumara, I. The Multifaceted Regulation of Mitochondrial Dynamics During Mitosis. Front. Cell Dev. Biol. 2021, 9, 767221. [Google Scholar] [CrossRef] [PubMed]
- Mitra, K.; Wunder, C.; Roysam, B.; Lin, G.; Lippincott-Schwartz, J. A Hyperfused Mitochondrial State Achieved at G1-S Regulates Cyclin E Buildup and Entry into S Phase. Proc. Natl. Acad. Sci. USA 2009, 106, 11960–11965. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Fan, M.; Candas, D.; Zhang, T.-Q.; Qin, L.; Eldridge, A.; Wachsmann-Hogiu, S.; Ahmed, K.M.; Chromy, B.A.; Nantajit, D.; et al. Cyclin B1/Cdk1 Coordinates Mitochondrial Respiration for Cell-Cycle G2/M Progression. Dev. Cell 2014, 29, 217–232. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Fan, M.; Candas, D.; Jiang, G.; Papadopoulos, S.; Tian, L.; Woloschak, G.; Grdina, D.J.; Li, J.J. CDK1 Enhances Mitochondrial Bioenergetics for Radiation-Induced DNA Repair. Cell Rep. 2015, 13, 2056–2063. [Google Scholar] [CrossRef] [PubMed]
- Kirova, D.G.; Judasova, K.; Vorhauser, J.; Zerjatke, T.; Leung, J.K.; Glauche, I.; Mansfeld, J. A ROS-Dependent Mechanism Promotes CDK2 Phosphorylation to Drive Progression through S Phase. Dev. Cell 2022, 57, 1712–1727.e9. [Google Scholar] [CrossRef]
- Santiappillai, N.T.; Abuhammad, S.; Slater, A.; Kirby, L.; McArthur, G.A.; Sheppard, K.E.; Smith, L.K. CDK4/6 Inhibition Reprograms Mitochondrial Metabolism in BRAFV600 Melanoma via a P53 Dependent Pathway. Cancers 2021, 13, 524. [Google Scholar] [CrossRef]
- Kashatus, D.F.; Lim, K.-H.; Brady, D.C.; Pershing, N.L.K.; Cox, A.D.; Counter, C.M. RALA and RALBP1 Regulate Mitochondrial Fission at Mitosis. Nat. Cell Biol. 2011, 13, 1108–1115. [Google Scholar] [CrossRef]
- Bertolin, G.; Bulteau, A.-L.; Alves-Guerra, M.-C.; Burel, A.; Lavault, M.-T.; Gavard, O.; Le Bras, S.; Gagné, J.-P.; Poirier, G.G.; Le Borgne, R.; et al. Aurora Kinase A Localises to Mitochondria to Control Organelle Dynamics and Energy Production. eLife 2018, 7, e38111. [Google Scholar] [CrossRef]
- Fernández Casafuz, A.B.; De Rossi, M.C.; Bruno, L. Mitochondrial Cellular Organization and Shape Fluctuations Are Differentially Modulated by Cytoskeletal Networks. Sci. Rep. 2023, 13, 4065. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.S.; Coscia, S.M.; Simpson, C.L.; Ortega, F.E.; Wait, E.C.; Heddleston, J.M.; Nirschl, J.J.; Obara, C.J.; Guedes-Dias, P.; Boecker, C.A.; et al. Actin Cables and Comet Tails Organize Mitochondrial Networks in Mitosis. Nature 2021, 591, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Kanfer, G.; Courthéoux, T.; Peterka, M.; Meier, S.; Soste, M.; Melnik, A.; Reis, K.; Aspenström, P.; Peter, M.; Picotti, P.; et al. Mitotic Redistribution of the Mitochondrial Network by Miro and Cenp-F. Nat. Commun. 2015, 6, 8015. [Google Scholar] [CrossRef] [PubMed]
- López-Doménech, G.; Covill-Cooke, C.; Ivankovic, D.; Halff, E.F.; Sheehan, D.F.; Norkett, R.; Birsa, N.; Kittler, J.T. Miro Proteins Coordinate Microtubule- and Actin-dependent Mitochondrial Transport and Distribution. EMBO J. 2018, 37, 321–336. [Google Scholar] [CrossRef] [PubMed]
- Donthamsetty, S.; Brahmbhatt, M.; Pannu, V.; Rida, P.; Ramarathinam, S.; Ogden, A.; Cheng, A.; Singh, K.K.; Aneja, R. Mitochondrial Genome Regulates Mitotic Fidelity by Maintaining Centrosomal Homeostasis. Cell Cycle 2014, 13, 2056–2255. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, P.; Yeon, B.; Wallis, S.S.; Godinho, S.A. Centrosome Amplification Fine Tunes Tubulin Acetylation to Differentially Control Intracellular Organization. EMBO J. 2023, 42, e112812. [Google Scholar] [CrossRef]
- Singla, V.; Reiter, J.F. The Primary Cilium as the Cell’s Antenna: Signaling at a Sensory Organelle. Science 2006, 313, 629–633. [Google Scholar] [CrossRef]
- Kasahara, K.; Inagaki, M. Primary Ciliary Signaling: Links with the Cell Cycle. Trends Cell Biol. 2021, 31, 954–964. [Google Scholar] [CrossRef]
- Bae, J.-E.; Kang, G.M.; Min, S.H.; Jo, D.S.; Jung, Y.-K.; Kim, K.; Kim, M.-S.; Cho, D.-H. Primary Cilia Mediate Mitochondrial Stress Responses to Promote Dopamine Neuron Survival in a Parkinson’s Disease Model. Cell Death Dis. 2019, 10, 952. [Google Scholar] [CrossRef]
- Burkhalter, M.D.; Sridhar, A.; Sampaio, P.; Jacinto, R.; Burczyk, M.S.; Donow, C.; Angenendt, M.; Competence Network for Congenital Heart Defects Investigators; Hempel, M.; Walther, P.; et al. Imbalanced Mitochondrial Function Provokes Heterotaxy via Aberrant Ciliogenesis. J. Clin. Investig. 2019, 129, 2841–2855. [Google Scholar] [CrossRef]
- Moruzzi, N.; Valladolid-Acebes, I.; Kannabiran, S.A.; Bulgaro, S.; Burtscher, I.; Leibiger, B.; Leibiger, I.B.; Berggren, P.-O.; Brismar, K. Mitochondrial Impairment and Intracellular Reactive Oxygen Species Alter Primary Cilia Morphology. Life Sci. Alliance 2022, 5, e202201505. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Lin, M.G.; Stowe, T.R.; Chen, S.; Zhu, M.; Stearns, T.; Franco, B.; Zhong, Q. Autophagy Promotes Primary Ciliogenesis by Removing OFD1 from Centriolar Satellites. Nature 2013, 502, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Pampliega, O.; Orhon, I.; Patel, B.; Sridhar, S.; Díaz-Carretero, A.; Beau, I.; Codogno, P.; Satir, B.H.; Satir, P.; Cuervo, A.M. Functional Interaction between Autophagy and Ciliogenesis. Nature 2013, 502, 194–200. [Google Scholar] [CrossRef]
- O’Flanagan, C.H.; Morais, V.A.; Wurst, W.; De Strooper, B.; O’Neill, C. The Parkinson’s Gene PINK1 Regulates Cell Cycle Progression and Promotes Cancer-Associated Phenotypes. Oncogene 2015, 34, 1363–1374. [Google Scholar] [CrossRef] [PubMed]
- Fry, A.M.; O’Regan, L.; Sabir, S.R.; Bayliss, R. Cell Cycle Regulation by the NEK Family of Protein Kinases. J. Cell Sci. 2012, 125, 4423–4433. [Google Scholar] [CrossRef] [PubMed]
- Fry, A.M. A Centrosomal Function for the Human Nek2 Protein Kinase, a Member of the NIMA Family of Cell Cycle Regulators. EMBO J. 1998, 17, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Fry, A.M. The Nek2 Protein Kinase: A Novel Regulator of Centrosome Structure. Oncogene 2002, 21, 6184–6194. [Google Scholar] [CrossRef] [PubMed]
- Faragher, A.J.; Fry, A.M. Nek2A Kinase Stimulates Centrosome Disjunction and Is Required for Formation of Bipolar Mitotic Spindles. MBoC 2003, 14, 2876–2889. [Google Scholar] [CrossRef]
- Prosser, S.L.; Sahota, N.K.; Pelletier, L.; Morrison, C.G.; Fry, A.M. Nek5 Promotes Centrosome Integrity in Interphase and Loss of Centrosome Cohesion in Mitosis. J. Cell Biol. 2015, 209, 339–348. [Google Scholar] [CrossRef]
- Belham, C.; Roig, J.; Caldwell, J.A.; Aoyama, Y.; Kemp, B.E.; Comb, M.; Avruch, J. A Mitotic Cascade of NIMA Family Kinases. J. Biol. Chem. 2003, 278, 34897–34909. [Google Scholar] [CrossRef]
- Bertran, M.T.; Sdelci, S.; Regué, L.; Avruch, J.; Caelles, C.; Roig, J. Nek9 Is a Plk1-Activated Kinase That Controls Early Centrosome Separation through Nek6/7 and Eg5: Plk1-Activated Nek9 Controls Centrosome Separation. EMBO J. 2011, 30, 2634–2647. [Google Scholar] [CrossRef]
- Du, J.; Cai, X.; Yao, J.; Ding, X.; Wu, Q.; Pei, S.; Jiang, K.; Zhang, Y.; Wang, W.; Shi, Y.; et al. The Mitotic Checkpoint Kinase NEK2A Regulates Kinetochore Microtubule Attachment Stability. Oncogene 2008, 27, 4107–4114. [Google Scholar] [CrossRef] [PubMed]
- White, M.C.; Quarmby, L.M. The NIMA-Family Kinase, Nek1 Affects the Stability of Centrosomes and Ciliogenesis. BMC Cell Biol. 2008, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Coene, K.L.M.; Mans, D.A.; Boldt, K.; Gloeckner, C.J.; van Reeuwijk, J.; Bolat, E.; Roosing, S.; Letteboer, S.J.F.; Peters, T.A.; Cremers, F.P.M.; et al. The Ciliopathy-Associated Protein Homologs RPGRIP1 and RPGRIP1L Are Linked to Cilium Integrity through Interaction with Nek4 Serine/Threonine Kinase. Hum. Mol. Genet. 2011, 20, 3592–3605. [Google Scholar] [CrossRef] [PubMed]
- Levedakou, E.N.; He, M.; Baptist, E.W.; Craven, R.J.; Cance, W.G.; Welcsh, P.L.; Simmons, A.; Naylor, S.L.; Leach, R.J.; Lewis, T.B. Two Novel Human Serine/Threonine Kinases with Homologies to the Cell Cycle Regulating Xenopus MO15, and NIMA Kinases: Cloning and Characterization of Their Expression Pattern. Oncogene 1994, 9, 1977–1988. [Google Scholar] [PubMed]
- Wang, W.; Cheng, X.; Lu, J.; Wei, J.; Fu, G.; Zhu, F.; Jia, C.; Zhou, L.; Xie, H.; Zheng, S. Mitofusin-2 Is a Novel Direct Target of P53. Biochem. Biophys. Res. Commun. 2010, 400, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Achanta, G.; Sasaki, R.; Feng, L.; Carew, J.S.; Lu, W.; Pelicano, H.; Keating, M.J.; Huang, P. Novel Role of P53 in Maintaining Mitochondrial Genetic Stability through Interaction with DNA Pol γ. EMBO J. 2005, 24, 3482–3492. [Google Scholar] [CrossRef]
- Tasdemir, E.; Maiuri, M.C.; Morselli, E.; Criollo, A.; D’Amelio, M.; Djavaheri-Mergny, M.; Cecconi, F.; Tavernarakis, N.; Kroemer, G. A Dual Role of P53 in the Control of Autophagy. Autophagy 2008, 4, 810–814. [Google Scholar] [CrossRef]
- Hoshino, A.; Mita, Y.; Okawa, Y.; Ariyoshi, M.; Iwai-Kanai, E.; Ueyama, T.; Ikeda, K.; Ogata, T.; Matoba, S. Cytosolic P53 Inhibits Parkin-Mediated Mitophagy and Promotes Mitochondrial Dysfunction in the Mouse Heart. Nat. Commun. 2013, 4, 2308. [Google Scholar] [CrossRef]
- Duan, L.; Perez, R.E.; Davaadelger, B.; Dedkova, E.N.; Blatter, L.A.; Maki, C.G. P53-Regulated Autophagy Is Controlled by Glycolysis and Determines Cell Fate. Oncotarget 2015, 6, 23135–23156. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basei, F.L.; e Silva, I.R.; Dias, P.R.F.; Ferezin, C.C.; Peres de Oliveira, A.; Issayama, L.K.; Moura, L.A.R.; da Silva, F.R.; Kobarg, J. The Mitochondrial Connection: The Nek Kinases’ New Functional Axis in Mitochondrial Homeostasis. Cells 2024, 13, 473. https://doi.org/10.3390/cells13060473
Basei FL, e Silva IR, Dias PRF, Ferezin CC, Peres de Oliveira A, Issayama LK, Moura LAR, da Silva FR, Kobarg J. The Mitochondrial Connection: The Nek Kinases’ New Functional Axis in Mitochondrial Homeostasis. Cells. 2024; 13(6):473. https://doi.org/10.3390/cells13060473
Chicago/Turabian StyleBasei, Fernanda L., Ivan Rosa e Silva, Pedro R. Firmino Dias, Camila C. Ferezin, Andressa Peres de Oliveira, Luidy K. Issayama, Livia A. R. Moura, Fernando Riback da Silva, and Jörg Kobarg. 2024. "The Mitochondrial Connection: The Nek Kinases’ New Functional Axis in Mitochondrial Homeostasis" Cells 13, no. 6: 473. https://doi.org/10.3390/cells13060473