
Virus Infection and Systemic Inflammation: Lessons Learnt from COVID-19 and Beyond

 , , and
, , and
Abstract
:1. Introduction
2. Crossing the Species Barrier—Human Infection with Zoonotic Viruses
3. From Local to Systemic—Disease Course and Immune Responses
4. Biomarkers for the Prediction of Disease Progression in COVID-19 and Infections with HPAIV
5. The Contribution of PRRs to the Innate Immune Responses during COVID-19 and HPAIV Infections
6. Adaptive Immune Response against SARS-CoV-2
7. Importance of Interferons for COVID-19
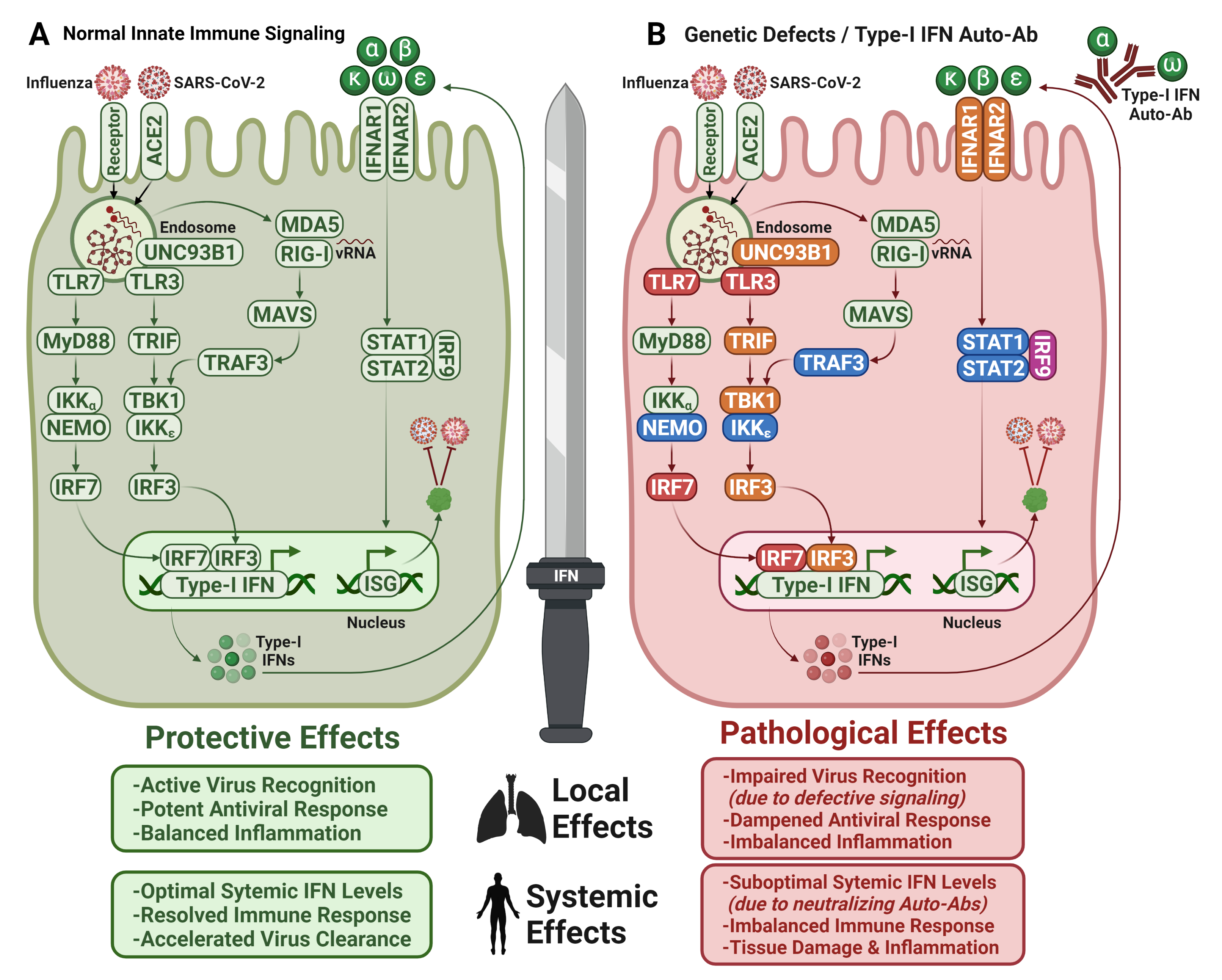
7.1. Type I IFN Autoantibodies in COVID-19

7.2. Therapeutic Application of IFNs for COVID-19 and HPAIV Infections
8. Antiviral and Immunomodulatory Treatments for COVID-19
8.1. Direct-Acting Antivirals for Treatment of COVID-19
8.2. Immunomodulatory Strategies to Alleviate Immunopathology in COVID-19
9. Long-Term Complications of COVID-19
10. COVID-19 and Long COVID in Children
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yu, H.; Peng, C.; Zhang, C.; Stoian, A.M.M.; Tazi, L.; Brennan, G.; Rothenburg, S. Maladaptation after a virus host switch leads to increased activation of the pro-inflammatory NF-κB pathway. Proc. Natl. Acad. Sci. USA 2022, 119, e2115354119. [Google Scholar] [CrossRef] [PubMed]
- Rouse, B.T.; Sehrawat, S. Immunity and immunopathology to viruses: What decides the outcome? Nat. Rev. Immunol. 2010, 10, 514–526. [Google Scholar] [CrossRef] [PubMed]
- Kalil, A.C.; Thomas, P.G. Influenza virus-related critical illness: Pathophysiology and epidemiology. Crit. Care 2019, 23, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandun, I.N.; Wibisono, H.; Sedyaningsih, E.R.; Yusharmen; Hadisoedarsuno, W.; Purba, W.; Santoso, H.; Septiawati, C.; Tresnaningsih, E.; Heriyanto, B.; et al. Three Indonesian Clusters of H5N1 Virus Infection in 2005. N. Engl. J. Med. 2006, 355, 2186–2194. [Google Scholar] [CrossRef] [Green Version]
- Koopmans, M.; Wilbrink, B.; Conyn, M.; Natrop, G.; van der Nat, H.; Vennema, H.; Meijer, A.; van Steenbergen, J.; Fouchier, R.; Osterhaus, A.; et al. Transmission of H7N7 avian influenza A virus to human beings during a large outbreak in commercial poultry farms in the Netherlands. Lancet 2004, 363, 587–593. [Google Scholar] [CrossRef]
- Olsen, S.J.; Ungchusak, K.; Sovann, L.; Uyeki, T.M.; Dowell, S.F.; Cox, N.J.; Aldis, W.; Chunsuttiwat, S. Family Clustering of Avian Influenza A (H5N1). Emerg. Infect. Dis. 2005, 11, 1799–1801. [Google Scholar] [CrossRef]
- Ungchusak, K.; Auewarakul, P.; Dowell, S.F.; Kitphati, R.; Auwanit, W.; Puthavathana, P.; Uiprasertkul, M.; Boonnak, K.; Pittayawonganon, C.; Cox, N.J.; et al. Probable Person-to-Person Transmission of Avian Influenza A (H5N1). New Engl. J. Med. 2005, 352, 333–340. [Google Scholar] [CrossRef] [Green Version]
- Hien, T.T.; Liem, N.T.; Dung, N.T.; San, L.T.; Mai, P.P.; Chau, N.V.V.; Suu, P.T.; Dong, V.C.; Mai, L.T.Q.; Thi, N.T.; et al. Avian Influenza A (H5N1) in 10 Patients in Vietnam. New Engl. J. Med. 2004, 350, 1179–1188. [Google Scholar] [CrossRef] [Green Version]
- Rogers, G.N.; Paulson, J.C. Receptor determinants of human and animal influenza virus isolates: Differences in receptor specificity of the H3 hemagglutinin based on species of origin. Virology 1983, 127, 361–373. [Google Scholar] [CrossRef]
- Stevens, J.; Blixt, O.; Glaser, L.; Taubenberger, J.K.; Palese, P.; Paulson, J.C.; Wilson, I.A. Glycan Microarray Analysis of the Hemagglutinins from Modern and Pandemic Influenza Viruses Reveals Different Receptor Specificities. J. Mol. Biol. 2006, 355, 1143–1155. [Google Scholar] [CrossRef]
- Nova, N. Cross-Species Transmission of Coronaviruses in Humans and Domestic Mammals, What Are the Ecological Mechanisms Driving Transmission, Spillover, and Disease Emergence? Front. Public Health 2021, 9, 717941. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Harder, T.; Koch, J.; Vygen-Bonnet, S.; Külper-Schiek, W.; Pilic, A.; Reda, S.; Scholz, S.; Wichmann, O. Efficacy and effectiveness of COVID-19 vaccines against SARS-CoV-2 infection: Interim results of a living systematic review, 1 January to 14 May 2021. Eurosurveillance 2021, 26, 2100563. [Google Scholar] [CrossRef]
- Remdesivir and three other drugs for hospitalised patients with COVID-19: Final results of the WHO Solidarity randomised trial and updated meta-analyses. Lancet 2022, 399, 1941–1953. [CrossRef]
- Cameroni, E.; Bowen, J.E.; Rosen, L.E.; Saliba, C.; Zepeda, S.K.; Culap, K.; Pinto, D.; VanBlargan, L.A.; De Marco, A.; di Iulio, J.; et al. Broadly neutralizing antibodies overcome SARS-CoV-2 Omicron antigenic shift. Nature 2022, 602, 664–670. [Google Scholar] [CrossRef]
- McCallum, M.; Czudnochowski, N.; Rosen, L.E.; Zepeda, S.K.; Bowen, J.E.; Walls, A.C.; Hauser, K.; Joshi, A.; Stewart, C.; Dillen, J.R.; et al. Structural basis of SARS-CoV-2 Omicron immune evasion and receptor engagement. Science 2022, 375, 864–868. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Addetia, A.; Hannon, W.W.; Choudhary, M.C.; Dingens, A.S.; Li, J.Z.; Bloom, J.D. Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. Science 2021, 371, 850–854. [Google Scholar] [CrossRef]
- Greaney, A.J.; Loes, A.N.; Crawford, K.H.; Starr, T.N.; Malone, K.D.; Chu, H.Y.; Bloom, J.D. Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe 2021, 29, 463–476.e6. [Google Scholar] [CrossRef]
- Murphy, K. SARS CoV-2 Detection From Upper and Lower Respiratory Tract Specimens. Chest 2020, 158, 1804–1805. [Google Scholar] [CrossRef]
- Nicholls, J.M.; Chan, M.C.W.; Chan, W.Y.; Wong, H.K.; Cheung, C.Y.; Kwong, D.L.W.; Wong, M.P.; Chui, W.H.; Poon, L.; Tsao, S.W.; et al. Tropism of avian influenza A (H5N1) in the upper and lower respiratory tract. Nat. Med. 2007, 13, 147–149. [Google Scholar] [CrossRef]
- Weinheimer, V.K.; Becher, A.; Tönnies, M.; Holland, G.; Knepper, J.; Bauer, T.T.; Schneider, P.; Neudecker, J.; Rückert, J.C.; Szymanski, K.; et al. Influenza A Viruses Target Type II Pneumocytes in the Human Lung. J. Infect. Dis. 2012, 206, 1685–1694. [Google Scholar] [CrossRef]
- Shinya, K.; Ebina, M.; Yamada, S.; Ono, M.; Kasai, N.; Kawaoka, Y. Influenza virus receptors in the human airway. Nature 2006, 440, 435–436. [Google Scholar] [CrossRef]
- Hou, Y.; Okuda, K.; Edwards, C.E.; Martinez, D.R.; Asakura, T.; Dinnon, K.H.; Kato, T.; Lee, R.E.; Yount, B.L.; Mascenik, T.M.; et al. SARS-CoV-2 Reverse Genetics Reveals a Variable Infection Gradient in the Respiratory Tract. Cell 2020, 182, 429–446.e14. [Google Scholar] [CrossRef]
- Ortiz, M.E.; Thurman, A.; Pezzulo, A.A.; Leidinger, M.R.; Klesney-Tait, J.A.; Karp, P.H.; Tan, P.; Wohlford-Lenane, C.; McCray, P.B.; Meyerholz, D.K. Heterogeneous expression of the SARS-Coronavirus-2 receptor ACE2 in the human respiratory tract. eBioMedicine 2020, 60, 102976. [Google Scholar] [CrossRef]
- Lakdawala, S.S.; Jayaraman, A.; Halpin, R.A.; Lamirande, E.W.; Shih, A.R.; Stockwell, T.B.; Lin, X.; Simenauer, A.; Hanson, C.T.; Vogel, L.; et al. The soft palate is an important site of adaptation for transmissible influenza viruses. Nature 2015, 526, 122–125. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.T.; Nakayama, T.; Wu, C.-T.; Goltsev, Y.; Jiang, S.; Gall, P.A.; Liao, C.-K.; Shih, L.-C.; Schürch, C.M.; McIlwain, D.R.; et al. ACE2 localizes to the respiratory cilia and is not increased by ACE inhibitors or ARBs. Nat. Commun. 2020, 11, 5453. [Google Scholar] [CrossRef]
- Lv, J.; Wang, Z.; Qu, Y.; Zhu, H.; Zhu, Q.; Tong, W.; Bao, L.; Lv, Q.; Cong, J.; Li, D.; et al. Distinct uptake, amplification, and release of SARS-CoV-2 by M1 and M2 alveolar macrophages. Cell Discov. 2021, 7, 24. [Google Scholar] [CrossRef]
- Chertow, D.; Stein, S.; Ramelli, S.; Grazioli, A.; Chung, J.-Y.; Singh, M.; Yinda, C.K.; Winkler, C.; Dickey, J.; Ylaya, K.; et al. SARS-CoV-2 Infection and Persistence Throughout the Human Body and Brain National Institutes of Health. 2021. Available online: https://www.researchsquare.com/article/rs-1139035/v1 (accessed on 18 May 2022).
- Flerlage, T.; Boyd, D.F.; Meliopoulos, V.; Thomas, P.G.; Schultz-Cherry, S. Influenza virus and SARS-CoV-2: Pathogenesis and host responses in the respiratory tract. Nat. Rev. Genet. 2021, 19, 425–441. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Lau, E.H.Y.; Wu, P.; Deng, X.; Wang, J.; Hao, X.; Lau, Y.C.; Wong, J.Y.; Guan, Y.; Tan, X.; et al. Temporal dynamics in viral shedding and transmissibility of COVID-19. Nat. Med. 2020, 26, 672–675. [Google Scholar] [CrossRef] [Green Version]
- Bi, Q.; Wu, Y.; Mei, S.; Ye, C.; Zou, X.; Zhang, Z.; Liu, X.; Wei, L.; Truelove, S.A.; Zhang, T.; et al. Epidemiology and transmission of COVID-19 in 391 cases and 1286 of their close contacts in Shenzhen, China: A retrospective cohort study. Lancet Infect. Dis. 2020, 20, 911–919. [Google Scholar] [CrossRef]
- Alimohamadi, Y.; Sepandi, M.; Taghdir, M.; Hosamirudsari, H. Determine the most common clinical symptoms in COVID-19 patients: A systematic review and meta-analysis. J. Prev. Med. Hyg. 2020, 61, E304–E312. [Google Scholar]
- Brann, D.H.; Tsukahara, T.; Weinreb, C.; Lipovsek, M.; Van Den Berge, K.; Gong, B.; Chance, R.; Macaulay, I.C.; Chou, H.-J.; Fletcher, R.B.; et al. Non-neuronal expression of SARS-CoV-2 entry genes in the olfactory system suggests mechanisms underlying COVID-19-associated anosmia. Sci. Adv. 2020, 6, eabc5801. [Google Scholar] [CrossRef]
- Burke, R.M.; Killerby, M.E.; Newton, S.; Ashworth, C.E.; Berns, A.L.; Brennan, S.; Bressler, J.M.; Bye, E.; Crawford, R.; Morano, L.H.; et al. Symptom Profiles of a Convenience Sample of Patients with COVID-19—United States, January–April 2020. MMWR. Morb. Mortal. Wkly. Rep. 2020, 69, 904–908. [Google Scholar] [CrossRef]
- Klaser, K.; Molteni, E.; Graham, M.S.; Canas, L.S.; Osterdahl, M.F.; Antonelli, M.; Chen, L.; Deng, J.; Murray, B.; Kerfoot, E.; et al. COVID-19 due to the B.1.617.2 (Delta) variant compared to B.1.1.7 (Alpha) variant of SARS-CoV-2: Two prospective observational cohort studies. medRxiv 2021. [Google Scholar] [CrossRef]
- Hagen, A. How Dangerous Is the Delta Variant (B.1.617.2)? Available online: https://asm.org/Articles/2021/July/How-Dangerous-is-the-Delta-Variant-B-1-617-2 (accessed on 20 June 2022).
- Shiehzadegan, S.; Alaghemand, N.; Fox, M.; Venketaraman, V. Analysis of the Delta Variant B.1.617.2 COVID-19. Clin. Pract. 2021, 11, 778–784. [Google Scholar] [CrossRef]
- Menni, C.; Valdes, A.M.; Polidori, L.; Antonelli, M.; Penamakuri, S.; Nogal, A.; Louca, P.; May, A.; Figueiredo, J.C.; Hu, C.; et al. Symptom prevalence, duration, and risk of hospital admission in individuals infected with SARS-CoV-2 during periods of omicron and delta variant dominance: A prospective observational study from the ZOE COVID Study. Lancet 2022, 399, 1618–1624. [Google Scholar] [CrossRef]
- Spiteri, G.; Fielding, J.; Diercke, M.; Campese, C.; Enouf, V.; Gaymard, A.; Bella, A.; Sognamiglio, P.; Moros, M.J.S.; Riutort, A.N.; et al. First cases of coronavirus disease 2019 (COVID-19) in the WHO European Region, 24 January to 21 February 2020. Eurosurveillance 2020, 25, 2000178. [Google Scholar] [CrossRef] [Green Version]
- Sinha, P.; Matthay, M.A.; Calfee, C.S. Is a “Cytokine Storm” Relevant to COVID-19? JAMA Intern. Med. 2020, 180, 1152–1154. [Google Scholar] [CrossRef]
- England, J.T.; Abdulla, A.; Biggs, C.; Lee, A.Y.; Hay, K.; Hoiland, R.L.; Wellington, C.L.; Sekhon, M.; Jamal, S.; Shojania, K.; et al. Weathering the COVID-19 storm: Lessons from hematologic cytokine syndromes. Blood Rev. 2021, 45, 100707. [Google Scholar] [CrossRef]
- Nadim, M.K.; Forni, L.G.; Mehta, R.L.; Connor, M.J., Jr.; Liu, K.D.; Ostermann, M.; Rimmelé, T.; Zarbock, A.; Bell, S.; Bihorac, A.; et al. COVID-19-associated acute kidney injury: Consensus report of the 25th Acute Disease Quality Initiative (ADQI) Workgroup. Nat. Rev. Nephrol. 2020, 16, 747–764. [Google Scholar] [CrossRef]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Robbins-Juarez, S.Y.; Qian, L.; King, K.L.; Stevens, J.S.; Husain, S.A.; Radhakrishnan, J.; Mohan, S. Outcomes for Patients With COVID-19 and Acute Kidney Injury: A Systematic Review and Meta-Analysis. Kidney Int. Rep. 2020, 5, 1149–1160. [Google Scholar] [CrossRef]
- Poissy, J.; Goutay, J.; Caplan, M.; Parmentier-Decrucq, E.; Duburcq, T.; Lassalle, F.; Jeanpierre, E.; Rauch, A.; Labreuche, J.; Susen, S.; et al. Pulmonary Embolism in Patients With COVID-19. Circulation 2020, 142, 184–186. [Google Scholar] [CrossRef]
- Ramos-Casals, M.; Brito-Zerón, P.; Mariette, X. Systemic and organ-specific immune-related manifestations of COVID-19. Nat. Rev. Rheumatol. 2021, 17, 315–332. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Reported Human Infections with Avian Influenza A V. Available online: https://www.cdc.gov/flu/avianflu/reported-human-infections.htm (accessed on 6 May 2022).
- Yang, L.; Zhao, X.; Li, X.; Bo, H.; Li, D.; Liu, J.; Wang, D. Case report for human infection with a highly pathogenic avian influenza A(H5N6) virus in Beijing, China 2019. Biosaf. Health 2020, 2, 49–52. [Google Scholar] [CrossRef]
- Yuen, K.-Y.; Chan, P.; Peiris, J.S.M.; Tsang, D.; Que, T.; Shortridge, K.; Cheung, P.; To, W.; Ho, E.; Sung, R.; et al. Clinical features and rapid viral diagnosis of human disease associated with avian influenza A H5N1 virus. Lancet 1998, 351, 467–471. [Google Scholar] [CrossRef]
- Yu, H.; Gao, Z.; Feng, Z.; Shu, Y.; Xiang, N.; Zhou, L.; Huai, Y.; Feng, L.; Peng, Z.; Li, Z.; et al. Clinical Characteristics of 26 Human Cases of Highly Pathogenic Avian Influenza A (H5N1) Virus Infection in China. PLoS ONE 2008, 3, e2985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koutsakos, M.; Kedzierska, K.; Subbarao, K. Immune Responses to Avian Influenza Viruses. J. Immunol. 2019, 202, 382–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- To, K.-F.; Chan, P.K.; Chan, K.-F.; Lee, W.-K.; Lam, W.-Y.; Wong, K.-F.; Tang, N.L.; Tsang, D.N.; Sung, R.Y.; Buckley, T.A.; et al. Pathology of fatal human infection associated with avian influenza A H5N1 virus. J. Med Virol. 2001, 63, 242–246. [Google Scholar] [CrossRef]
- Le, M.T.Q.; Wertheim, H.F.L.; Nguyen, H.D.; Taylor, W.; Hoang, P.V.M.; Vuong, C.D.; Nguyen, H.L.K.; Nguyen, H.H.; Nguyen, T.Q.; Nguyen, T.V.; et al. Influenza A H5N1 Clade 2.3.4 Virus with a Different Antiviral Susceptibility Profile Replaced Clade 1 Virus in Humans in Northern Vietnam. PLoS ONE 2008, 3, e3339. [Google Scholar] [CrossRef]
- Kandun, I.N.; Tresnaningsih, E.; Purba, W.H.; Lee, V.; Samaan, G.; Harun, S.; Soni, E.; Septiawati, C.; Setiawati, T.; Sariwati, E.; et al. Factors associated with case fatality of human H5N1 virus infections in Indonesia: A case series. Lancet 2008, 372, 744–749. [Google Scholar] [CrossRef]
- Oner, A.F.; Bay, A.; Arslan, S.; Akdeniz, H.; Sahin, H.A.; Cesur, Y.; Epcacan, S.; Yilmaz, N.; Deger, I.; Kizilyildiz, B.; et al. Avian Influenza A (H5N1) Infection in Eastern Turkey in 2006. N. Engl. J. Med. 2006, 355, 2179–2185. [Google Scholar] [CrossRef]
- Chotpitayasunondh, T.; Ungchusak, K.; Hanshaoworakul, W.; Chunsuthiwat, S.; Sawanpanyalert, P.; Kijphati, R.; Lochindarat, S.; Srisan, P.; Suwan, P.; Osotthanakorn, Y.; et al. Human Disease from Influenza A (H5N1), Thailand. Emerg. Infect. Dis. 2005, 11, 201–209. [Google Scholar] [CrossRef]
- Buchy, P.; Mardy, S.; Vong, S.; Toyoda, T.; Aubin, J.-T.; Miller, M.; Touch, S.; Sovann, L.; Dufourcq, J.-B.; Richner, B.; et al. Influenza A/H5N1 virus infection in humans in Cambodia. J. Clin. Virol. 2007, 39, 164–168. [Google Scholar] [CrossRef]
- Uyeki, T.M. Human Infection with Highly Pathogenic Avian Influenza A (H5N1) Virus: Review of Clinical Issues. Clin. Infect. Dis. 2009, 49, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Pan, M.; Gao, R.; Lv, Q.; Huang, S.; Zhou, Z.; Yang, L.; Li, X.; Zhao, X.; Zou, X.; Tong, W.; et al. Human infection with a novel, highly pathogenic avian influenza A (H5N6) virus: Virological and clinical findings. J. Infect. 2016, 72, 52–59. [Google Scholar] [CrossRef]
- Chen, H.; Yuan, H.; Gao, R.; Zhang, J.; Wang, D.; Xiong, Y.; Fan, G.; Yang, F.; Li, X.; Zhou, J.; et al. Clinical and epidemiological characteristics of a fatal case of avian influenza A H10N8 virus infection: A descriptive study. Lancet 2014, 383, 714–721. [Google Scholar] [CrossRef]
- Peiris, J.; Yu, W.; Leung, C.; Cheung, C.; Ng, W.; Nicholls, J.; Ng, T.; Chan, K.; Lai, S.; Lim, W.; et al. Re-emergence of fatal human influenza A subtype H5N1 disease. Lancet 2004, 363, 617–619. [Google Scholar] [CrossRef]
- De Jong, M.D.; Simmons, C.P.; Thanh, T.T.; Hien, V.M.; Smith, G.J.D.; Chau, T.N.B.; Hoang, D.M.; Chau, N.V.V.; Khanh, T.H.; Dong, V.C.; et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat. Med. 2006, 12, 1203–1207. [Google Scholar] [CrossRef]
- Chan, M.C.; Cheung, C.Y.; Chui, W.H.; Tsao, S.W.; Nicholls, J.M.; Chan, Y.O.; Chan, R.W.; Long, H.T.; Poon, L.L.; Guan, Y.; et al. Proinflammatory cytokine responses induced by influenza A (H5N1) viruses in primary human alveolar and bronchial epithelial cells. Respir. Res. 2005, 6, 135. [Google Scholar] [CrossRef] [Green Version]
- Krischuns, T.; Günl, F.; Henschel, L.; Binder, M.; Willemsen, J.; Schloer, S.; Rescher, U.; Gerlt, V.; Zimmer, G.; Nordhoff, C.; et al. Phosphorylation of TRIM28 Enhances the Expression of IFN-β and Proinflammatory Cytokines During HPAIV Infection of Human Lung Epithelial Cells. Front. Immunol. 2018, 9, 2229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, C.; Poon, L.; Lau, A.; Luk, W.; Lau, Y.; Shortridge, K.; Gordon, S.; Guan, Y.; Peiris, J. Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: A mechanism for the unusual severity of human disease? Lancet 2002, 360, 1831–1837. [Google Scholar] [CrossRef]
- Lipatov, A.S.; Andreansky, S.; Webby, R.J.; Hulse, D.J.; Rehg, J.E.; Krauss, S.; Perez, D.R.; Doherty, P.C.; Webster, R.G.; Sangster, M.Y. Pathogenesis of Hong Kong H5N1 influenza virus NS gene reassortants in mice: The role of cytokines and B- and T-cell responses. J. Gen. Virol. 2005, 86, 1121–1130. [Google Scholar] [CrossRef] [PubMed]
- Sakabe, S.; Iwatsuki-Horimoto, K.; Takano, R.; Nidom, C.A.; Le, M.T.Q.; Nagamura-Inoue, T.; Horimoto, T.; Yamashita, N.; Kawaoka, Y. Cytokine production by primary human macrophages infected with highly pathogenic H5N1 or pandemic H1N1 2009 influenza viruses. J. Gen. Virol. 2011, 92, 1428–1434. [Google Scholar] [CrossRef] [PubMed]
- Mok, K.P.; Wong, C.H.K.; Cheung, C.Y.; Chan, M.C.W.; Lee, S.M.Y.; Nicholls, J.M.; Guan, Y.; Peiris, J.S.M. Viral Genetic Determinants of H5N1 Influenza Viruses That Contribute to Cytokine Dysregulation. J. Infect. Dis. 2009, 200, 1104–1112. [Google Scholar] [CrossRef] [PubMed]
- Salomon, R.; Hoffmann, E.; Webster, R.G. Inhibition of the cytokine response does not protect against lethal H5N1 influenza infection. Proc. Natl. Acad. Sci. USA 2007, 104, 12479–12481. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Qiao, J.; Zhao, L.; He, G.; Li, K.; Wang, J.; Tian, Y.; Wang, H. Effect of dexamethasone on acute respiratory distress syndrome induced by the H5N1 virus in mice. Eur. Respir. J. 2009, 33, 852–860. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.K.; Rosenheim, J.; Bell, L.C.; Chandran, A.; Guerra-Assuncao, J.A.; Pollara, G.; Whelan, M.; Artico, J.; Joy, G.; Kurdi, H.; et al. Blood transcriptional biomarkers of acute viral infection for detection of pre-symptomatic SARS-CoV-2 infection: A nested, case-control diagnostic accuracy study. Lancet Microbe 2021, 2, e508–e517. [Google Scholar] [CrossRef]
- Tang, B.M.; Shojaei, M.; Parnell, G.P.; Huang, S.; Nalos, M.; Teoh, S.; O’Connor, K.; Schibeci, S.; Phu, A.L.; Kumar, A.; et al. A novel immune biomarker IFI27 discriminates between influenza and bacteria in patients with suspected respiratory infection. Eur. Respir. J. 2017, 49, 1602098. [Google Scholar] [CrossRef] [Green Version]
- Bellmann-Weiler, R.; Lanser, L.; Barket, R.; Rangger, L.; Schapfl, A.; Schaber, M.; Fritsche, G.; Wöll, E.; Weiss, G. Prevalence and Predictive Value of Anemia and Dysregulated Iron Homeostasis in Patients with COVID-19 Infection. J. Clin. Med. 2020, 9, 2429. [Google Scholar] [CrossRef]
- Terpos, E.; Ntanasis-Stathopoulos, I.; Elalamy, I.; Kastritis, E.; Sergentanis, T.N.; Politou, M.; Psaltopoulou, T.; Gerotziafas, G.; Dimopoulos, M.A. Hematological findings and complications of COVID-19. Am. J. Hematol. 2020, 95, 834–847. [Google Scholar] [CrossRef] [Green Version]
- Huang, I.; Pranata, R. Lymphopenia in severe coronavirus disease-2019 (COVID-19): Systematic review and meta-analysis. J. Intensive Care 2020, 8, 36. [Google Scholar] [CrossRef]
- Huang, I.; Pranata, R.; Lim, M.A.; Oehadian, A.; Alisjahbana, B. C-reactive protein, procalcitonin, D-dimer, and ferritin in severe coronavirus disease-2019: A meta-analysis. Ther. Adv. Respir. Dis. 2020, 14, 1753466620937175. [Google Scholar] [CrossRef]
- Smilowitz, N.R.; Kunichoff, D.; Garshick, M.; Shah, B.; Pillinger, M.; Hochman, J.S.; Berger, J.S. C-reactive protein and clinical outcomes in patients with COVID-19. Eur. Hearth J. 2021, 42, 2270–2279. [Google Scholar] [CrossRef]
- Hu, R.; Han, C.; Pei, S.; Yin, M.; Chen, X. Procalcitonin levels in COVID-19 patients. Int. J. Antimicrob. Agents 2020, 56, 106051. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, J.; Yang, Y.; Ma, H.; Li, Z.; Zhang, J.; Cheng, J.; Zhang, X.; Zhao, Y.; Xia, Z.; et al. The role of interleukin-6 in monitoring severe case of coronavirus disease 2019. EMBO Mol. Med. 2020, 12, e12421. [Google Scholar] [CrossRef]
- Chen, H.; Wang, J.; Su, N.; Bao, X.; Li, Y.; Jin, J. Simplified immune-dysregulation index: A novel marker predicts 28-day mortality of intensive care patients with COVID-19. Intensiv. Care Med. 2020, 46, 1645–1647. [Google Scholar] [CrossRef]
- Giannakodimos, I.; Gkountana, G.-V.; Lykouras, D.; Karkoulias, K.; Tsakas, S. The Role of Interleukin-6 in the Pathogenesis, Prognosis and Treatment of Severe COVID-19. Curr. Med. Chem. 2020, 28, 5328–5338. [Google Scholar] [CrossRef]
- Liu, F.; Li, L.; Xu, M.; Wu, J.; Luo, D.; Zhu, Y.; Li, B.; Song, X.; Zhou, X. Prognostic value of interleukin-6, C-reactive protein, and procalcitonin in patients with COVID-19. J. Clin. Virol. 2020, 127, 104370. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of Immune Response in Patients With Coronavirus 2019 (COVID-19) in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef]
- Zhang, L.; Yan, X.; Fan, Q.; Liu, H.; Liu, X.; Liu, Z.; Zhang, Z. D-dimer levels on admission to predict in-hospital mortality in patients with Covid-19. J. Thromb. Haemost. 2020, 18, 1324–1329. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Cao, J.; Wang, Q.; Shi, Q.; Liu, K.; Luo, Z.; Chen, X.; Chen, S.; Yu, K.; Huang, Z.; et al. D-dimer as a biomarker for disease severity and mortality in COVID-19 patients: A case control study. J. Intensiv. Care 2020, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Rovas, A.; Osiaevi, I.; Buscher, K.; Sackarnd, J.; Tepasse, P.-R.; Fobker, M.; Kühn, J.; Braune, S.; Göbel, U.; Thölking, G.; et al. Microvascular dysfunction in COVID-19: The MYSTIC study. Angiogenesis 2021, 24, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Vasileva, D.; Badawi, A. C-reactive protein as a biomarker of severe H1N1 influenza. Agents Actions 2019, 68, 39–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beigel, J.H.; Farrar, J.; Han, A.M.; Hayden, F.G.; Hyer, R.; de Jong, M.D.; Lochindarat, S.; Nguyen, T.K.T.; Nguyen, T.H.; Tran, T.H.; et al. Avian influenza A (H5N1) infection in humans. N. Engl. J. Med. 2005, 353, 1374–1385. [Google Scholar]
- Soepandi, P.Z.; Burhan, E.; Mangunnegoro, H.; Nawas, A.; Aditama, T.Y.; Partakusuma, L.; Isbaniah, F.; Ikhsan, M.; Swidarmoko, B.; Sutiyoso, A.; et al. Clinical Course of Avian Influenza A(H5N1) in Patients at the Persahabatan Hospital, Jakarta, Indonesia, 2005. Chest 2010, 138, 665–673. [Google Scholar] [CrossRef]
- Diamond, M.S.; Kanneganti, T.-D. Innate immunity: The first line of defense against SARS-CoV-2. Nat. Immunol. 2022, 23, 165–176. [Google Scholar] [CrossRef]
- Ramos, I.; Fernandez-Sesma, A. Innate Immunity to H5N1 Influenza Viruses in Humans. Viruses 2012, 4, 3363–3388. [Google Scholar] [CrossRef] [Green Version]
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern Recognition Receptors and the Innate Immune Response to Viral Infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef] [Green Version]
- Kanneganti, T. Intracellular innate immune receptors: Life inside the cell. Immunol. Rev. 2020, 297, 5–12. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [Green Version]
- Lian, Q.; Zhang, K.; Zhang, Z.; Duan, F.; Guo, L.; Luo, W.; Mok, B.W.-Y.; Thakur, A.; Ke, X.; Motallebnejad, P.; et al. Differential effects of macrophage subtypes on SARS-CoV-2 infection in a human pluripotent stem cell-derived model. Nat. Commun. 2022, 13, 2028. [Google Scholar] [CrossRef]
- Dalskov, L.; Møhlenberg, M.; Thyrsted, J.; Blay-Cadanet, J.; Poulsen, E.T.; Folkersen, B.H.; Skaarup, S.H.; Olagnier, D.; Reinert, L.; Enghild, J.J.; et al. SARS-CoV-2 evades immune detection in alveolar macrophages. EMBO Rep. 2020, 21, e51252. [Google Scholar] [CrossRef]
- Wang, J.; Nikrad, M.P.; Travanty, E.A.; Zhou, B.; Phang, T.; Gao, B.; Alford, T.; Ito, Y.; Nahreini, P.; Hartshorn, K.; et al. Innate Immune Response of Human Alveolar Macrophages during Influenza A Infection. PLoS ONE 2012, 7, e29879. [Google Scholar] [CrossRef] [Green Version]
- Cuevas, A.M.; Clark, J.M.; Potter, J.J. Increased TLR/MyD88 signaling in patients with obesity: Is there a link to COVID-19 disease severity? Int. J. Obes. 2021, 45, 1152–1154. [Google Scholar] [CrossRef]
- Sohn, K.M.; Lee, S.-G.; Kim, H.J.; Cheon, S.; Jeong, H.; Lee, J.; Kim, I.S.; Silwal, P.; Kim, Y.J.; Paik, S.; et al. COVID-19 Patients Upregulate Toll-like Receptor 4-mediated Inflammatory Signaling That Mimics Bacterial Sepsis. J. Korean Med Sci. 2020, 35, e343. [Google Scholar] [CrossRef]
- Zhao, Y.; Kuang, M.; Li, J.; Zhu, L.; Jia, Z.; Guo, X.; You, F. Publisher Correction: SARS-CoV-2 spike protein interacts with and activates TLR4 (Cell Research, (2021), 31, 7, (818-820), 10.1038/s41422-021-00495-9). Cell Res. 2021, 31, 825. [Google Scholar] [CrossRef]
- Yamada, T.; Sato, S.; Sotoyama, Y.; Orba, Y.; Sawa, H.; Yamauchi, H.; Sasaki, M.; Takaoka, A. RIG-I triggers a signaling-abortive anti-SARS-CoV-2 defense in human lung cells. Nat. Immunol. 2021, 22, 820–828. [Google Scholar] [CrossRef]
- Hui, K.P.Y.; Lee, S.M.Y.; Cheung, C.-Y.; Mao, H.; Lai, A.K.W.; Chan, R.W.Y.; Chan, M.C.W.; Tu, W.; Guan, Y.; Lau, Y.-L.; et al. H5N1 Influenza Virus–Induced Mediators Upregulate RIG-I in Uninfected Cells by Paracrine Effects Contributing to Amplified Cytokine Cascades. J. Infect. Dis. 2011, 204, 1866–1878. [Google Scholar] [CrossRef] [Green Version]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef]
- Yin, X.; Riva, L.; Pu, Y.; Martin-Sancho, L.; Kanamune, J.; Yamamoto, Y.; Sakai, K.; Gotoh, S.; Miorin, L.; De Jesus, P.D.; et al. MDA5 Governs the Innate Immune Response to SARS-CoV-2 in Lung Epithelial Cells. Cell Rep. 2021, 34, 108628. [Google Scholar] [CrossRef]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- Te Velthuis, A.J.W.; Long, J.C.; Bauer, D.L.V.; Fan, R.L.Y.; Yen, H.-L.; Sharps, J.; Siegers, J.Y.; Killip, M.J.; French, H.; Oliva-Martín, M.J.; et al. Mini viral RNAs act as innate immune agonists during influenza virus infection. Nat. Microbiol. 2018, 3, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Arai, Y.; Yamanaka, I.; Okomato, T.; Isobe, A.; Nakai, N.; Kamimura, N.; Suzuki, T.; Daidoji, T.; Ono, T.; et al. Stimulation of Dysregulated IFN- β Responses by Aberrant SARS-CoV-2 Small Viral RNAs Acting as RIG-I Agonists. 2022. Available online: https://assets.researchsquare.com/files/rs-1271053/v2/b9e39a7d-71a8-46e9-b293-e1334e366176.pdf?c=1647456052 (accessed on 6 May 2022).
- Zheng, M.; Karki, R.; Williams, E.P.; Yang, D.; Fitzpatrick, E.; Vogel, P.; Jonsson, C.B.; Kanneganti, T.-D. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat. Immunol. 2021, 22, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Shafiei, M.S.; Longoria, C.; Schoggins, J.W.; Savani, R.C.; Zaki, H. SARS-CoV-2 spike protein induces inflammation via TLR2-dependent activation of the NF-κB pathway. eLife 2021, 10, e68563. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Pillai, P.S. Innate immunity to influenza virus infection. Nat. Rev. Immunol. 2014, 14, 315–328. [Google Scholar] [CrossRef]
- Dey, M.; Cao, C.; Dar, A.C.; Tamura, T.; Ozato, K.; Sicheri, F.; Dever, T.E. Mechanistic Link between PKR Dimerization, Autophosphorylation, and eIF2α Substrate Recognition. Cell 2005, 122, 901–913. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Renner, D.M.; Comar, C.E.; Whelan, J.N.; Reyes, H.M.; Cardenas-Diaz, F.L.; Truitt, R.; Tan, L.H.; Dong, B.; Alysandratos, K.D.; et al. SARS-CoV-2 induces double-stranded RNA-mediated innate immune responses in respiratory epithelial-derived cells and cardiomyocytes. Proc. Natl. Acad. Sci. USA 2021, 118, e2022643118. [Google Scholar] [CrossRef]
- Sette, A.; Crotty, S. Adaptive immunity to SARS-CoV-2 and COVID-19. Cell 2021, 184, 861–880. [Google Scholar] [CrossRef]
- Chen, G.; Wu, D.; Guo, W.; Cao, Y.; Huang, D.; Wang, H.; Wang, T.; Zhang, X.; Chen, H.; Yu, H.; et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Investig. 2020, 130, 2620–2629. [Google Scholar] [CrossRef] [Green Version]
- Oja, A.E.; Saris, A.; Ghandour, C.A.; Kragten, N.A.; Hogema, B.M.; Nossent, E.J.; Heunks, L.M.; Cuvalay, S.; Slot, E.; Linty, F.; et al. Divergent SARS-CoV-2-specific T- and B-cell responses in severe but not mild COVID-19 patients. Eur. J. Immunol. 2020, 50, 1998–2012. [Google Scholar] [CrossRef]
- Diao, B.; Wang, C.; Tan, Y.; Chen, X.; Liu, Y.; Ning, L.; Chen, L.; Li, M.; Liu, Y.; Wang, G.; et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19). Front. Immunol. 2020, 11, 827. [Google Scholar] [CrossRef]
- Mathew, D.; Giles, J.R.; Baxter, A.E.; Oldridge, D.A.; Greenplate, A.R.; Wu, J.E.; Alanio, C.; Kuri-Cervantes, L.; Pampena, M.B.; D’Andrea, K.; et al. Deep immune profiling of COVID-19 patients reveals distinct immunotypes with therapeutic implications. Science 2020, 369, 6508. [Google Scholar] [CrossRef]
- Moss, P. The T cell immune response against SARS-CoV-2. Nat. Immunol. 2022, 23, 186–193. [Google Scholar] [CrossRef]
- Hall, V.J.; Foulkes, S.; Charlett, A.; Atti, A.; Monk, E.J.M.; Simmons, R.; Wellington, E.; Cole, M.J.; Saei, A.; Oguti, B.; et al. SARS-CoV-2 infection rates of antibody-positive compared with antibody-negative health-care workers in England: A large, multicentre, prospective cohort study (SIREN). Lancet 2021, 397, 1459–1469. [Google Scholar] [CrossRef]
- Galbraith, M.D.; Kinning, K.T.; Sullivan, K.D.; Araya, P.; Smith, K.P.; Granrath, R.E.; Shaw, J.R.; Baxter, R.; Jordan, K.R.; Russell, S.; et al. Specialized interferon action in COVID-19. Proc. Natl. Acad. Sci. USA 2022, 119, e2116730119. [Google Scholar] [CrossRef]
- Meyts, I.; Casanova, J. Viral infections in humans and mice with genetic deficiencies of the type I IFN response pathway. Eur. J. Immunol. 2021, 51, 1039–1061. [Google Scholar] [CrossRef]
- Ciancanelli, M.J.; Huang, S.X.L.; Luthra, P.; Garner, H.; Itan, Y.; Volpi, S.; Lafaille, F.G.; Trouillet, C.; Schmolke, M.; Albrecht, R.A.; et al. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science 2015, 348, 448–453. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, N.; Melki, I.; Jing, H.; Habib, T.; Huang, S.S.; Danielson, J.; Kula, T.; Drutman, S.; Belkaya, S.; Rattina, V.; et al. Life-threatening influenza pneumonitis in a child with inherited IRF9 deficiency. J. Exp. Med. 2018, 215, 2567–2585. [Google Scholar] [CrossRef]
- Lim, H.K.; Huang, S.X.; Chen, J.; Kerner, G.; Gilliaux, O.; Bastard, P.; Dobbs, K.; Hernandez, N.; Goudin, N.; Hasek, M.L.; et al. Severe influenza pneumonitis in children with inherited TLR3 deficiency. J. Exp. Med. 2019, 216, 2038–2056. [Google Scholar] [CrossRef]
- Bastard, P.; Zhang, Q.; Zhang, S.-Y.; Jouanguy, E.; Casanova, J.-L. Type I interferons and SARS-CoV-2: From cells to organisms. Curr. Opin. Immunol. 2022, 74, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.D.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570. [Google Scholar] [CrossRef]
- Zhang, S.-Y.; Zhang, Q.; Casanova, J.-L.; Su, H.C.; Abel, L.; Bastard, P.; Cobat, A.; Jouanguy, E.; Notarangelo, L.; Covid The COVID Team. Severe COVID-19 in the young and healthy: Monogenic inborn errors of immunity? Nat. Rev. Immunol. 2020, 20, 455–456. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Puel, A.; Zhang, S.; Eidenschenk, C.; Ku, C.-L.; Casrouge, A.; Picard, C.; von Bernuth, H.; Senechal, B.; Plancoulaine, S.; et al. Human TLR-7-, -8-, and -9-Mediated Induction of IFN-α/β and -λ Is IRAK-4 Dependent and Redundant for Protective Immunity to Viruses. Immunity 2005, 23, 465–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barreiro, L.B.; Ben-Ali, M.; Quach, H.; Laval, G.; Patin, E.; Pickrell, J.K.; Bouchier, C.; Tichit, M.; Neyrolles, O.; Gicquel, B.; et al. Evolutionary Dynamics of Human Toll-Like Receptors and Their Different Contributions to Host Defense. PLoS Genet. 2009, 5, e1000562. [Google Scholar] [CrossRef] [Green Version]
- Puel, A.; Bastard, P.; Bustamante, J.; Casanova, J.L. Human autoantibodies underlying infectious diseases. J. Exp. Med. 2022, 219, e20211387. [Google Scholar] [CrossRef] [PubMed]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.-H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Béziat, V.; et al. Auto-antibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020, 370, eabd4585. [Google Scholar] [CrossRef]
- Abers, M.S.; Rosen, L.B.; Delmonte, O.M.; Shaw, E.; Bastard, P.; Imberti, L.; Quaresima, V.; Biondi, A.; Bonfanti, P.; Castagnoli, R.; et al. Neutralizing type-I interferon autoantibodies are associated with delayed viral clearance and intensive care unit admission in patients with COVID-19. Immunol. Cell Biol. 2021, 99, 917–921. [Google Scholar] [CrossRef]
- Acosta-Ampudia, Y.; Monsalve, D.M.; Rojas, M.; Rodríguez, Y.; Gallo, J.E.; Salazar-Uribe, J.C.; Santander, M.J.; Cala, M.P.; Zapata, W.; Zapata, M.I.; et al. COVID-19 convalescent plasma composition and immunological effects in severe patients. J. Autoimmun. 2021, 118, 102598. [Google Scholar] [CrossRef]
- Vazquez, S.E.; Bastard, P.; Kelly, K.; Gervais, A.; Norris, P.J.; Dumont, L.J.; Casanova, J.-L.; Anderson, M.S.; DeRisi, J.L. Neutralizing Autoantibodies to Type I Interferons in COVID-19 Convalescent Donor Plasma. J. Clin. Immunol. 2021, 41, 1169–1171. [Google Scholar] [CrossRef]
- Wang, E.Y.; Mao, T.; Klein, J.; Dai, Y.; Huck, J.D.; Jaycox, J.R.; Liu, F.; Zhou, T.; Israelow, B.; Wong, P.; et al. Diverse Functional Autoantibodies in Patients with COVID-19. Nature 2021, 595, 283–288. [Google Scholar] [CrossRef]
- Ziegler, C.G.; Miao, V.N.; Owings, A.H.; Navia, A.W.; Tang, Y.; Bromley, J.D.; Lotfy, P.; Sloan, M.; Laird, H.; Williams, H.B.; et al. Impaired local intrinsic immunity to SARS-CoV-2 infection in severe COVID-19. Cell 2021, 184, 4713–4733.e22. [Google Scholar] [CrossRef]
- Troya, J.; Bastard, P.; Planas-Serra, L.; Ryan, P.; Ruiz, M.; de Carranza, M.; Torres, J.; Martínez, A.; Abel, L.; Casanova, J.-L.; et al. Neutralizing Autoantibodies to Type I IFNs in >10% of Patients with Severe COVID-19 Pneumonia Hospitalized in Madrid, Spain. J. Clin. Immunol. 2021, 41, 914–922. [Google Scholar] [CrossRef]
- Zhang, Q.; Bastard, P.; Karbuz, A.; Gervais, A.; Tayoun, A.A.; Aiuti, A.; Belot, A.; Bolze, A.; Gaudet, A.; Bondarenko, A.; et al. Human genetic and immunological determinants of critical COVID-19 pneumonia. Nature 2022, 603, 587–598. [Google Scholar] [CrossRef]
- Carapito, R.; Li, R.; Helms, J.; Carapito, C.; Gujja, S.; Rolli, V.; Guimaraes, R.; Malagon-Lopez, J.; Spinnhirny, P.; Lederle, A.; et al. Identification of driver genes for critical forms of COVID-19 in a deeply phenotyped young patient cohort. Sci. Transl. Med. 2022, 14, abj7521. [Google Scholar] [CrossRef]
- Chang, S.E.; Feng, A.; Meng, W.; Apostolidis, S.A.; Mack, E.; Artandi, M.; Barman, L.; Bennett, K.; Chakraborty, S.; Chang, I.; et al. New-onset IgG autoantibodies in hospitalized patients with COVID-19. Nat. Commun. 2021, 12, 5417. [Google Scholar] [CrossRef]
- Chauvineau-Grenier, A.; Bastard, P.; Servajean, A.; Gervais, A.; Rosain, J.; Jouanguy, E.; Cobat, A.; Casanova, J.-L.; Rossi, B. Autoantibodies Neutralizing Type I Interferons in 20% of COVID-19 Deaths in a French Hospital. J. Clin. Immunol. 2022, 42, 459–470. [Google Scholar] [CrossRef]
- Goncalves, D.; Mezidi, M.; Bastard, P.; Perret, M.; Saker, K.; Fabien, N.; Pescarmona, R.; Lombard, C.; Walzer, T.; Casanova, J.; et al. Antibodies against type I interferon: Detection and association with severe clinical outcome in COVID-19 patients. Clin. Transl. Immunol. 2021, 10, e1327. [Google Scholar] [CrossRef]
- Koning, R.; Bastard, P.; Casanova, J.-L.; Brouwer, M.C.; van de Beek, D.; van Agtmael, M.; Algera, A.G.; Appelman, B.; van Baarle, F.; Bax, D.; et al. Autoantibodies against type I interferons are associated with multi-organ failure in COVID-19 patients. Intensiv. Care Med. 2021, 47, 704–706. [Google Scholar] [CrossRef]
- Raadsen, M.P.; Gharbharan, A.; Jordans, C.C.E.; Mykytyn, A.Z.; Lamers, M.M.; van den Doel, P.B.; Endeman, H.; van den Akker, J.P.C.; Geurtsvan Kessel, C.H.; Koopmans, M.P.G.; et al. Interferon-α2 Auto-antibodies in Convalescent Plasma Therapy for COVID-19. J. Clin. Immunol. 2022, 42, 232–239. [Google Scholar] [CrossRef]
- Solanich, X.; Rigo-Bonnin, R.; Gumucio, V.-D.; Bastard, P.; Rosain, J.; Philippot, Q.; Perez-Fernandez, X.-L.; Fuset-Cabanes, M.-P.; Gordillo-Benitez, M.; Suarez-Cuartin, G.; et al. Pre-existing Autoantibodies Neutralizing High Concentrations of Type I Interferons in Almost 10% of COVID-19 Patients Admitted to Intensive Care in Barcelona. J. Clin. Immunol. 2021, 41, 1733–1744. [Google Scholar] [CrossRef] [PubMed]
- Van der Wijst, M.G.P.; Vazquez, S.E.; Hartoularos, G.C.; Bastard, P.; Grant, T.; Bueno, R.; Lee, D.S.; Greenland, J.R.; Sun, Y.; Perez, R.; et al. Type I interferon autoantibodies are associated with systemic immune alterations in patients with COVID-19. Sci. Transl. Med. 2021, 13, eabh2624. [Google Scholar] [CrossRef] [PubMed]
- Trouillet-Assant, S.; Viel, S.; Gaymard, A.; Pons, S.; Richard, J.-C.; Perret, M.; Villard, M.; Brengel-Pesce, K.; Lina, B.; Mezidi, M.; et al. Type I IFN immunoprofiling in COVID-19 patients. J. Allergy Clin. Immunol. 2020, 146, 206–208.e2. [Google Scholar] [CrossRef] [PubMed]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Credle, J.J.; Gunn, J.; Sangkhapreecha, P.; Monaco, D.R.; Zheng, X.A.; Tsai, H.J.; Wilbon, A.; Morgenlander, W.R.; Dong, Y.; Jayaraman, S.; et al. Neutralizing IFNL3 Autoantibodies in Severe COVID-19 Identified Using Molecular Indexing of Proteins by Self-Assembly. bioRxiv 2021. [Google Scholar] [CrossRef]
- Beccuti, G.; Ghizzoni, L.; Cambria, V.; Codullo, V.; Sacchi, P.; Lovati, E.; Mongodi, S.; Iotti, G.A.; Mojoli, F. A COVID-19 pneumonia case report of autoimmune polyendocrine syndrome type 1 in Lombardy, Italy: Letter to the editor. J. Endocrinol. Investig. 2020, 43, 1175–1177. [Google Scholar] [CrossRef]
- Carpino, A.; Buganza, R.; Matarazzo, P.; Tuli, G.; Pinon, M.; Calvo, P.L.; Montin, D.; Licciardi, F.; De Sanctis, L. Autoimmune Polyendocrinopathy–Candidiasis–Ectodermal Dystrophy in Two Siblings: Same Mutations but Very Different Phenotypes. Genes 2021, 12, 169. [Google Scholar] [CrossRef]
- Bastard, P.; Orlova, E.; Sozaeva, L.; Lévy, R.; James, A.; Schmitt, M.M.; Ochoa, S.; Kareva, M.; Rodina, Y.; Gervais, A.; et al. Preexisting autoantibodies to type I IFNs underlie critical COVID-19 pneumonia in patients with APS-1. J. Exp. Med. 2021, 218, e20210554. [Google Scholar] [CrossRef]
- Meisel, C.; Akbil, B.; Meyer, T.; Lankes, E.; Corman, V.M.; Staudacher, O.; Unterwalder, N.; Kölsch, U.; Drosten, C.; Mall, M.A.; et al. Mild COVID-19 despite autoantibodies against type I IFNs in autoimmune polyendocrine syndrome type. J. Clin. Investig. 2021, 131, e150867. [Google Scholar] [CrossRef]
- Lopez, J.; Mommert, M.; Mouton, W.; Pizzorno, A.; Brengel-Pesce, K.; Mezidi, M.; Villard, M.; Lina, B.; Richard, J.-C.; Fassier, J.-B.; et al. Early nasal type I IFN immunity against SARS-CoV-2 is compromised in patients with autoantibodies against type I IFNs. J. Exp. Med. 2021, 218, e20211211. [Google Scholar] [CrossRef]
- Bastard, P.; Michailidis, E.; Hoffmann, H.-H.; Chbihi, M.; Le Voyer, T.; Rosain, J.; Philippot, Q.; Seeleuthner, Y.; Gervais, A.; Materna, M.; et al. Auto-antibodies to type I IFNs can underlie adverse reactions to yellow fever live attenuated vaccine. J. Exp. Med. 2021, 218, e20202486. [Google Scholar] [CrossRef]
- Klein, S.L.; Pekosz, A.; Park, H.-S.; Ursin, R.L.; Shapiro, J.R.; Benner, S.E.; Littlefield, K.; Kumar, S.; Naik, H.M.; Betenbaugh, M.J.; et al. Sex, age, and hospitalization drive antibody responses in a COVID-19 convalescent plasma donor population. J. Clin. Investig. 2020, 130, 6141–6150. [Google Scholar] [CrossRef]
- Wang, T.T.; Ravetch, J.V. Functional diversification of IgGs through Fc glycosylation. J. Clin. Investig. 2019, 129, 3492–3498. [Google Scholar] [CrossRef] [Green Version]
- Beck, D.B.; Aksentijevich, I. Susceptibility to severe COVID-19. Science 2020, 370, 404–405. [Google Scholar] [CrossRef]
- Paul, F.; Pellegrini, S.; Uzé, G. IFNA2: The prototypic human alpha interferon. Gene 2015, 567, 132–137. [Google Scholar] [CrossRef] [Green Version]
- Severa, M.; Farina, C.; Salvetti, M.; Coccia, E.M. Three Decades of Interferon-β in Multiple Sclerosis: Can We Repurpose This Information for the Management of SARS-CoV2 Infection? Front. Immunol. 2020, 11, 1459. [Google Scholar] [CrossRef]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Reder, A.T.; Feng, X. How Type I Interferons Work in Multiple Sclerosis and Other Diseases: Some Unexpected Mechanisms. J. Interf. Cytokine Res. 2014, 34, 589–599. [Google Scholar] [CrossRef] [Green Version]
- Mantlo, E.; Bukreyeva, N.; Maruyama, J.; Paessler, S.; Huang, C. Antiviral activities of type I interferons to SARS-CoV-2 infection. Antivir. Res. 2020, 179, 104811. [Google Scholar] [CrossRef]
- Kumar, S.; Çalışkan, D.M.; Janowski, J.; Faist, A.; Conrad, B.C.G.; Lange, J.; Ludwig, S.; Brunotte, L. Beyond Vaccines: Clinical Status of Prospective COVID-19 Therapeutics. Front. Immunol. 2021, 12, 752227. [Google Scholar] [CrossRef]
- Stanifer, M.L.; Guo, C.; Doldan, P.; Boulant, S. Importance of Type I and III Interferons at Respiratory and Intestinal Barrier Surfaces. Front. Immunol. 2020, 11, 608645. [Google Scholar] [CrossRef] [PubMed]
- Vanderheiden, A.; Ralfs, P.; Chirkova, T.; Upadhyay, A.A.; Zimmerman, M.G.; Bedoya, S.; Aoued, H.; Tharp, G.M.; Pellegrini, K.L.; Manfredi, C.; et al. Type I and Type III Interferons Restrict SARS-CoV-2 Infection of Human Airway Epithelial Cultures. J. Virol. 2020, 94, e00985-20. [Google Scholar] [CrossRef] [PubMed]
- Feld, J.J.; Kandel, C.; Biondi, M.J.; Kozak, R.A.; Zahoor, M.A.; Lemieux, C.; Borgia, S.M.; Boggild, A.K.; Powis, J.; McCready, J.; et al. Peginterferon lambda for the treatment of outpatients with COVID-19: A phase 2, placebo-controlled randomised trial. Lancet Respir. Med. 2021, 9, 498–510. [Google Scholar] [CrossRef]
- Metz-Zumaran, C.; Kee, C.; Doldan, P.; Guo, C.; Stanifer, M.L.; Boulant, S. Increased Sensitivity of SARS-CoV-2 to Type III Interferon in Human Intestinal Epithelial Cells. J. Virol. 2022, 96, e0170521. [Google Scholar] [CrossRef] [PubMed]
- Stanifer, M.L.; Kee, C.; Cortese, M.; Zumaran, C.M.; Triana, S.; Mukenhirn, M.; Kraeusslich, H.-G.; Alexandrov, T.; Bartenschlager, R.; Boulant, S. Critical Role of Type III Interferon in Controlling SARS-CoV-2 Infection in Human Intestinal Epithelial Cells. Cell Rep. 2020, 32, 107863. [Google Scholar] [CrossRef] [PubMed]
- EIGER Biopharmaceuticals. Eiger’s Single-Dose Peginterferon Lambda for COVID-19 Reduced Risk of Hospitalization or ER Visits by 50% in a Predominantly Vaccinated Population in Phase 3 TOGETHER Study. Available online: https://www.prnewswire.com/news-releases/eigers-single-dose-peginterferon-lambda-for-covid-19-reduced-risk-of-hospitalization-or-er-visits-by-50-in-a-predominantly-vaccinated-population-in-phase-3-together-study-301504827.html (accessed on 4 May 2022).
- Haasbach, E.; Droebner, K.; Vogel, A.B.; Planz, O. Low-Dose Interferon Type I Treatment Is Effective Against H5N1 and Swine-Origin H1N1 Influenza A Viruses In Vitro and In Vivo. J. Interf. Cytokine Res. 2011, 31, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Bennett, A.L.; Smith, D.W.; Cummins, M.J.; Jacoby, P.A.; Cummins, J.M.; Beilharz, M.W. Low-dose oral interferon alpha as prophylaxis against viral respiratory illness: A double-blind, parallel controlled trial during an influenza pandemic year. Influ. Other Respir. Viruses 2013, 7, 854–862. [Google Scholar] [CrossRef]
- Sutter, K.; Dickow, J.; Dittmer, U. Interferon α subtypes in HIV infection. Cytokine Growth Factor Rev. 2018, 40, 13–18. [Google Scholar] [CrossRef]
- Matos, A.D.R.; Wunderlich, K.; Schloer, S.; Schughart, K.; Geffers, R.; Seders, M.; De Witt, M.; Christersson, A.; Wiewrodt, R.; Wiebe, K.; et al. Antiviral potential of human IFN-α subtypes against influenza A H3N2 infection in human lung explants reveals subtype-specific activities. Emerg. Microbes Infect. 2019, 8, 1763–1776. [Google Scholar] [CrossRef] [Green Version]
- Guo, K.; Shen, G.; Kibbie, J.; Gonzalez, T.; Dillon, S.M.; Smith, H.A.; Cooper, E.H.; Lavender, K.; Hasenkrug, K.J.; Sutter, K.; et al. Qualitative Differences Between the IFNα subtypes and IFNβ Influence Chronic Mucosal HIV-1 Pathogenesis. PLOS Pathog. 2020, 16, e1008986. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Lai, F.; Wang, Y.; Sutter, K.; Dittmer, U.; Ye, J.; Zai, W.; Liu, M.; Shen, F.; et al. Functional Comparison of Interferon-α Subtypes Reveals Potent Hepatitis B Virus Suppression by a Concerted Action of Interferon-α and Interferon-γ Signaling. Hepatology 2021, 73, 486–502. [Google Scholar] [CrossRef]
- Schuhenn, J.; Meister, T.L.; Todt, D.; Bracht, T.; Schork, K.; Billaud, J.-N.; Elsner, C.; Heinen, N.; Karakoese, Z.; Haid, S.; et al. Differential interferon-α subtype induced immune signatures are associated with suppression of SARS-CoV-2 infection. Proc. Natl. Acad. Sci. USA 2022, 119, e2111600119. [Google Scholar] [CrossRef] [PubMed]
- Hung, I.F.N.; To, K.; Lee, C.-K.; Lee, K.-L.; Chan, K.K.C.; Yan, W.-W.; Liu, R.; Watt, C.-L.; Chan, W.-M.; Lai, K.-Y.; et al. Convalescent Plasma Treatment Reduced Mortality in Patients With Severe Pandemic Influenza A (H1N1) 2009 Virus Infection. Clin. Infect. Dis. 2011, 52, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhou, J.; Huang, Y.; Liu, X.; Xu, Y.; Chen, S.; Liu, D.; Lin, Z.; Liu, X.; Li, Y. Efficacy of convalescent plasma for the treatment of severe influenza. Crit. Care 2020, 24, 469. [Google Scholar] [CrossRef]
- Marano, G.; Vaglio, S.; Pupella, S.; Facco, G.; Catalano, L.; Liumbruno, G.M.; Grazzini, G. Convalescent plasma: New evidence for an old therapeutic tool? Blood Transfus. 2016, 14, 152–157. [Google Scholar] [CrossRef] [Green Version]
- Nitesh, J.; Kashyap, R.; Surani, S.R. What we learned in the past year in managing our COVID-19 patients in intensive care units? World J. Crit. Care Med. 2021, 10, 81–101. [Google Scholar] [CrossRef]
- Brown, B.L.; McCullough, J. Treatment for emerging viruses: Convalescent plasma and COVID-19. Transfus. Apher. Sci. 2020, 59, 102790. [Google Scholar] [CrossRef]
- Bertoglio, F.; Meier, D.; Langreder, N.; Steinke, S.; Rand, U.; Simonelli, L.; Heine, P.A.; Ballmann, R.; Schneider, K.-T.; Roth, K.D.R.; et al. SARS-CoV-2 neutralizing human recombinant antibodies selected from pre-pandemic healthy donors binding at RBD-ACE2 interface. Nat. Commun. 2021, 12, 1577. [Google Scholar] [CrossRef]
- Gavriatopoulou, M.; Ntanasis-Stathopoulos, I.; Korompoki, E.; Fotiou, D.; Migkou, M.; Tzanninis, I.-G.; Psaltopoulou, T.; Kastritis, E.; Terpos, E.; Dimopoulos, M.A. Emerging treatment strategies for COVID-19 infection. Clin. Exp. Med. 2021, 21, 167–179. [Google Scholar] [CrossRef]
- Majumder, J.; Minko, T. Recent Developments on Therapeutic and Diagnostic Approaches for COVID-19. AAPS J. 2021, 23, 14. [Google Scholar] [CrossRef] [PubMed]
- National Institutes of Health. COVID-19 Treatment Guidelines; Anti-SARS-CoV-2 Antibody Products. Available online: https://www.covid19treatmentguidelines.nih.gov/ (accessed on 4 May 2022).
- Ader, F.; Bouscambert-Duchamp, M.; Hites, M.; Peiffer-Smadja, N.; Poissy, J.; Belhadi, D.; Diallo, A.; Lê, M.-P.; Peytavin, G.; Staub, T.; et al. Remdesivir plus standard of care versus standard of care alone for the treatment of patients admitted to hospital with COVID-19 (DisCoVeRy): A phase 3, randomised, controlled, open-label trial. Lancet Infect. Dis. 2022, 22, 209–221. [Google Scholar] [CrossRef]
- Williamson, B.N.; Feldmann, F.; Schwarz, B.; Meade-White, K.; Porter, D.P.; Schulz, J.; Van Doremalen, N.; Leighton, I.; Yinda, C.K.; Pérez-Pérez, L.; et al. Clinical benefit of remdesivir in rhesus macaques infected with SARS-CoV-2. Nature 2020, 585, 273–276. [Google Scholar] [CrossRef]
- Tanni, S.E.; Silvinato, A.; Floriano, I.; Bacha, H.A.; Barbosa, A.N.; Bernardo, W.M. Use of remdesivir in patients with COVID-19: A systematic review and meta-analysis. J. Bras. Pneumol. 2022, 48, e20210393. [Google Scholar] [CrossRef]
- Mulangu, S.; Dodd, L.E.; Davey, R.T., Jr.; Tshiani Mbaya, O.; Proschan, M.; Mukadi, D.; Lusakibanza Manzo, M.; Nzolo, D.; Tshomba Oloma, A.; Ibanda, A.; et al. A Randomized, Controlled Trial of Ebola Virus Disease Therapeutics. N. Engl. J. Med. 2019, 381, 2293–2303. [Google Scholar] [CrossRef]
- Cho, A.; Saunders, O.L.; Butler, T.; Zhang, L.; Xu, J.; Vela, J.E.; Feng, J.Y.; Ray, A.S.; Kim, C.U. Synthesis and antiviral activity of a series of 1′-substituted 4-aza-7,9-dideazaadenosine C-nucleosides. Bioorganic Med. Chem. Lett. 2012, 22, 2705–2707. [Google Scholar] [CrossRef]
- Lo, M.K.; Jordan, R.; Arvey, A.; Sudhamsu, J.; Shrivastava-Ranjan, P.; Hotard, A.L.; Flint, M.; McMullan, L.; Siegel, D.; Clarke, M.O.; et al. GS-5734 and its parent nucleoside analog inhibit Filo-, Pneumo-, and Paramyxoviruses. Sci. Rep. 2017, 7, srep43395. [Google Scholar] [CrossRef]
- U.S. Food & Drug Administration. FDA Takes Actions to Expand Use of Treatment for Outpatients with Mild-to-Moderate COVID-19. Available online: https://www.fda.gov/news-events/press-announcements/fda-takes-actions-expand-use-treatment-outpatients-mild-moderate-covid-19 (accessed on 4 May 2022).
- Zhou, S.; Hill, C.S.; Sarkar, S.; Tse, L.V.; Woodburn, B.M.D.; Schinazi, R.F.; Sheahan, T.P.; Baric, R.S.; Heise, M.T.; Swanstrom, R. β-d-N4-hydroxycytidine Inhibits SARS-CoV-2 Through Lethal Mutagenesis But Is Also Mutagenic To Mammalian Cells. J. Infect. Dis. 2021, 224, 415–419. [Google Scholar] [CrossRef]
- Furuta, Y.; Gowen, B.B.; Takahashi, K.; Shiraki, K.; Smee, D.F.; Barnard, D.L. Favipiravir (T-705), a novel viral RNA polymerase inhibitor. Antivir. Res. 2013, 100, 446–454. [Google Scholar] [CrossRef] [Green Version]
- Duwe, S. Influenza viruses—Antiviral therapy and resistance. GMS Infect. Dis. 2017, 5, Doc04. [Google Scholar] [CrossRef]
- Sidwell, R.W.; Barnard, D.L.; Day, C.W.; Smee, D.F.; Bailey, K.W.; Wong, M.-H.; Morrey, J.D.; Furuta, Y. Efficacy of Orally Administered T-705 on Lethal Avian Influenza A (H5N1) Virus Infections in Mice. Antimicrob. Agents Chemother. 2007, 51, 845–851. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Zhang, Y.; Huang, J.; Yin, P.; Cheng, Z.; Wu, J.; Chen, S.; Zhang, Y.; Chen, B.; Lu, M.; et al. Favipiravir Versus Arbidol for Clinical Recovery Rate in Moderate and Severe Adult COVID-19 Patients: A Prospective, Multicenter, Open-Label, Randomized Controlled Clinical Trial. Front. Pharmacol. 2021, 12, 683296. [Google Scholar] [CrossRef] [PubMed]
- Bosaeed, M.; Alharbi, A.; Mahmoud, E.; Alrehily, S.; Bahlaq, M.; Gaifer, Z.; Alturkistani, H.; Alhagan, K.; Alshahrani, S.; Tolbah, A.; et al. Efficacy of favipiravir in adults with mild COVID-19: A randomized, double-blind, multicentre, placebo-controlled clinical trial. Clin. Microbiol. Infect. 2022, 28, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.O.; Jin, P.; Rahman, K.M. Strategies for drug repurposing against coronavirus targets. Curr. Res. Pharmacol. Drug Discov. 2022, 3, 100072. [Google Scholar] [CrossRef] [PubMed]
- Extance, A. Covid-19: What is the evidence for the antiviral Paxlovid? BMJ 2022, 377, o1037. [Google Scholar] [CrossRef]
- Lamb, Y.N. Nirmatrelvir Plus Ritonavir: First Approval. Drugs 2022, 82, 585–591. [Google Scholar] [CrossRef]
- Hung, Y.-P.; Lee, J.-C.; Chiu, C.-W.; Lee, C.-C.; Tsai, P.-J.; Hsu, I.-L.; Ko, W.-C. Oral Nirmatrelvir/Ritonavir Therapy for COVID-19: The Dawn in the Dark? Antibiotics 2022, 11, 220. [Google Scholar] [CrossRef]
- Azkur, A.K.; Akdis, M.; Azkur, D.; Sokolowska, M.; Van De Veen, W.; Brüggen, M.-C.; O’Mahony, L.; Gao, Y.; Nadeau, K.; Akdis, C.A. Immune response to SARS-CoV-2 and mechanisms of immunopathological changes in COVID-19. Allergy 2020, 75, 1564–1581. [Google Scholar] [CrossRef]
- Rizk, J.G.; Kalantar-Zadeh, K.; Mehra, M.R.; Lavie, C.J.; Rizk, Y.; Forthal, D.N. Pharmaco-Immunomodulatory Therapy in COVID-19. Drugs 2020, 80, 1267–1292. [Google Scholar] [CrossRef]
- Kennedy, G.A.; Varelias, A.; Vuckovic, S.; Le Texier, L.; Gartlan, K.H.; Zhang, P.; Thomas, G.; Anderson, L.; Boyle, G.; Cloonan, N.; et al. Addition of interleukin-6 inhibition with tocilizumab to standard graft-versus-host disease prophylaxis after allogeneic stem-cell transplantation: A phase 1/2 trial. Lancet Oncol. 2014, 15, 1451–1459. [Google Scholar] [CrossRef]
- Sheng, F.S.F.; Han, M.H.M.; Huang, Z.H.Z.; Zhang, L.Z.L. Interleukin 6 receptor inhibitor tocilizumab suppresses cytokine expression, inflammasome activation and phagocytosis in a cell model of sepsis. Pharmazie 2016, 71, 636–639. [Google Scholar] [CrossRef]
- Abani, O.; Abbas, A.; Abbas, F.; Abbas, M.; Abbasi, S.; Abbass, H.; Abbott, A.; Abdallah, N.; Abdelaziz, A.; Abdelfattah, M.; et al. Tocilizumab in patients admitted to hospital with COVID-19 (RECOVERY): A randomised, controlled, open-label, platform trial. Lancet 2021, 397, 1637–1645. [Google Scholar] [CrossRef]
- Alzghari, S.K.; Acuña, V.S. Supportive Treatment with Tocilizumab for COVID-19: A Systematic Review. J. Clin. Virol. 2020, 127, 104380. [Google Scholar] [CrossRef]
- Morris, G.; Bortolasci, C.C.; Puri, B.K.; Marx, W.; O’Neil, A.; Athan, E.; Walder, K.; Berk, M.; Olive, L.; Carvalho, A.F.; et al. The cytokine storms of COVID-19, H1N1 influenza, CRS and MAS compared. Can one sized treatment fit all? Cytokine 2021, 144, 155593. [Google Scholar] [CrossRef]
- Kyriazopoulou, E.; Huet, T.; Cavalli, G.; Gori, A.; Kyprianou, M.; Pickkers, P.; Eugen-Olsen, J.; Clerici, M.; Veas, F.; Chatellier, G.; et al. Effect of anakinra on mortality in patients with COVID-19: A systematic review and patient-level meta-analysis. Lancet Rheumatol. 2021, 3, e690–e697. [Google Scholar] [CrossRef]
- Kyriazopoulou, E.; Poulakou, G.; Milionis, H.; Metallidis, S.; Adamis, G.; Tsiakos, K.; Fragkou, A.; Rapti, A.; Damoulari, C.; Fantoni, M.; et al. Early treatment of COVID-19 with anakinra guided by soluble urokinase plasminogen receptor plasma levels: A double-blind, randomized controlled phase 3 trial. Nat. Med. 2021, 27, 1752–1760. [Google Scholar] [CrossRef]
- Feldmann, M.; Maini, R.N.; Woody, J.N.; Holgate, S.T.; Winter, G.; Rowland, M.; Richards, D.; Hussell, T. Trials of anti-tumour necrosis factor therapy for COVID-19 are urgently needed. Lancet 2020, 395, 1407–1409. [Google Scholar] [CrossRef]
- Okeke, F.; Mone, A.; Swaminath, A. The Course of SARS-COV2 Infection Was Not Severe in a Crohn’s Patient Who Administered Maintenance Anti-TNF Therapy Overlapping the Early Pre-Symptomatic Period of Infection. Antibodies 2020, 9, 42. [Google Scholar] [CrossRef]
- Duret, P.-M.; Sebbag, E.; Mallick, A.; Gravier, S.; Spielmann, L.; Messer, L. Recovery from COVID-19 in a patient with spondyloarthritis treated with TNF-alpha inhibitor etanercept. Ann. Rheum. Dis. 2020, 79, 1251–1252. [Google Scholar] [CrossRef]
- Abdullah, A.; Neurath, M.F.; Atreya, R. Mild COVID-19 Symptoms in an Infliximab-Treated Ulcerative Colitis Patient: Can Ongoing Anti-TNF Therapy Protect against the Viral Hyperinflammatory Response and Avoid Aggravated Outcomes? Visc. Med. 2020, 36, 338–342. [Google Scholar] [CrossRef]
- Darwish, I.; Mubareka, S.; Liles, W.C. Immunomodulatory therapy for severe influenza. Expert Rev. Anti-infective Ther. 2011, 9, 807–822. [Google Scholar] [CrossRef]
- Guo, Y.; Hu, K.; Li, Y.; Lu, C.; Ling, K.; Cai, C.; Wang, W.; Ye, D. Targeting TNF-α for COVID-19: Recent Advanced and Controversies. Front. Public Health 2022, 10, 833967. [Google Scholar] [CrossRef] [PubMed]
- Alijotas-Reig, J.; Esteve-Valverde, E.; Belizna, C.; Selva-O’Callaghan, A.; Pardos-Gea, J.; Quintana, A.; Mekinian, A.; Anunciacion-Llunell, A.; Miro-Mur, F.A. Immunomodulatory therapy for the management of severe COVID-Beyond the anti-viral therapy: A comprehensive review. Autoimmun. Rev. 2020, 19, 102569. [Google Scholar] [CrossRef]
- Richardson, P.; Griffin, I.; Tucker, C.; Smith, D.; Oechsle, O.; Phelan, A.; Rawling, M.; Savory, E.; Stebbing, J. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet 2020, 395, e30–e31. [Google Scholar] [CrossRef] [Green Version]
- Safaei, S.; Karimi-Googheri, M. Letter to the Editor: Toll-Like Receptor Antagonists as a Potential Therapeutic Strategy Against Cytokine Storm in COVID-19-Infected Patients. Viral Immunol. 2021, 34, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Manik, M.; Singh, R.K. Role of toll-like receptors in modulation of cytokine storm signaling in SARS-CoV-2-induced COVID-19. J. Med Virol. 2022, 94, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Patra, R.; Das, N.C.; Mukherjee, S. Targeting human TLRs to combat COVID-19: A solution? J. Med Virol. 2021, 93, 615–617. [Google Scholar] [CrossRef] [PubMed]
- Perrin-Cocon, L.; Aublin-Gex, A.; Sestito, S.E.; Shirey, K.A.; Patel, M.C.; André, P.; Blanco, J.; Vogel, S.N.; Peri, F.; Lotteau, V. TLR4 antagonist FP7 inhibits LPS-induced cytokine production and glycolytic reprogramming in dendritic cells, and protects mice from lethal influenza infection. Sci. Rep. 2017, 7, srep40791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, C. Focus shifts to antibody cocktails for COVID-19 cytokine storm. Nat. Biotechnol. 2020, 38, 905–908. [Google Scholar] [CrossRef]
- Feuillet, V.; Canard, B.; Trautmann, A. Combining Antivirals and Immunomodulators to Fight COVID-19. Trends Immunol. 2021, 42, 31–44. [Google Scholar] [CrossRef]
- Liu, Q.; Zhou, Y.-H.; Yang, Z.-Q. The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell. Mol. Immunol. 2016, 13, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Z.; Liu, J.; Shi, D.; Chen, W.; Li, J.; Yan, R.; Bi, Y.; Hu, W.; Zhu, Z.; Yu, Y.; et al. Glucocorticoids improve severe or critical COVID-19 by activating ACE2 and reducing IL-6 levels. Int. J. Biol. Sci. 2020, 16, 2382–2391. [Google Scholar] [CrossRef]
- Keller, M.J.; Kitsis, E.A.; Arora, S.; Chen, J.-T.; Agarwal, S.; Ross, M.J.; Tomer, Y.; Southern, W. Effect of Systemic Glucocorticoids on Mortality or Mechanical Ventilation in Patients With COVID-19. J. Hosp. Med. 2020, 15, 489–493. [Google Scholar] [CrossRef]
- Honigsbaum, M.; Krishnan, L. Taking pandemic sequelae seriously: From the Russian influenza to COVID-19 long-haulers. Lancet 2020, 396, 1389–1391. [Google Scholar] [CrossRef]
- Sellers, S.A.; Hagan, R.S.; Hayden, F.G.; Fischer, W.A. The hidden burden of influenza: A review of the extra-pulmonary complications of influenza infection. Influ. Other Respir. Viruses 2017, 11, 372–393. [Google Scholar] [CrossRef]
- Kumar, K.; Guirgis, M.; Zieroth, S.; Lo, E.; Menkis, A.H.; Arora, R.C.; Freed, D.H. Influenza Myocarditis and Myositis: Case Presentation and Review of the Literature. Can. J. Cardiol. 2011, 27, 514–522. [Google Scholar] [CrossRef]
- Aranda, J.; Oriol, I.; Martín, M.; Feria, L.; Vázquez, N.; Rhyman, N.; Vall-Llosera, E.; Pallarés, N.; Coloma, A.; Pestaña, M.; et al. Long-term impact of COVID-19 associated acute respiratory distress syndrome. J. Infect. 2021, 83, 581–588. [Google Scholar] [CrossRef]
- Chopra, V.; Flanders, S.A.; O’Malley, M.; Malani, A.N.; Prescott, H.C. Sixty-Day Outcomes Among Patients Hospitalized With COVID-19. Ann. Intern. Med. 2021, 174, 576–578. [Google Scholar] [CrossRef]
- Martin-Villares, C.; Molina-Ramirez, C.P.; Bartolome-Benito, M.; Bernal-Sprekelsen, M.; Perez-Fernandez, A.; Alcantara-Armenteros, S.; Monjas-Cánovas, I.; Sancho-Mestre, M.; Alemán-Lopez, O.; Deola-Trasserra, M.D.; et al. Outcome of 1890 tracheostomies for critical COVID-19 patients: A national cohort study in Spain. Eur. Arch. Oto-Rhino-Laryngol. 2021, 278, 1605–1612. [Google Scholar] [CrossRef]
- Zhao, Y.-M.; Shang, Y.-M.; Song, W.-B.; Li, Q.-Q.; Xie, H.; Xu, Q.-F.; Jia, J.-L.; Li, L.-M.; Mao, H.-L.; Zhou, X.-M.; et al. Follow-up study of the pulmonary function and related physiological characteristics of COVID-19 survivors three months after recovery. eClinicalMedicine 2020, 25, 100463. [Google Scholar] [CrossRef]
- Shaw, B.; Daskareh, M.; Gholamrezanezhad, A. The lingering manifestations of COVID-19 during and after convalescence: Update on long-term pulmonary consequences of coronavirus disease 2019 (COVID-19). La Radiol. Med. 2021, 126, 40–46. [Google Scholar] [CrossRef]
- Van Gassel, R.J.J.; Bels, J.L.M.; Raafs, A.; van Bussel, B.C.T.; van de Poll, M.C.G.; Simons, S.O.; van der Meer, L.W.L.; Gietema, H.A.; Posthuma, R.; van Santen, S. High Prevalence of Pulmonary Sequelae at 3 Months after Hospital Discharge in Mechanically Ventilated Survivors of COVID-19. Am. J. Respir. Crit. Care Med. 2021, 203, 371–374. [Google Scholar] [CrossRef]
- Huang, C.; Huang, L.; Wang, Y.; Li, X.; Ren, L.; Gu, X.; Kang, L.; Guo, L.; Liu, M.; Zhou, X.; et al. 6-month consequences of COVID-19 in patients discharged from hospital: A cohort study. Lancet 2021, 397, 220–232. [Google Scholar] [CrossRef]
- Méndez, R.; Latorre, A.; González-Jiménez, P.; Feced, L.; Bouzas, L.; Yépez, K.; Ferrando, A.; Zaldívar-Olmeda, E.; Reyes, S.; Menéndez, R. Reduced Diffusion Capacity in COVID-19 Survivors. Ann. Am. Thorac. Soc. 2021, 18, 1253–1255. [Google Scholar] [CrossRef]
- Carfì, A.; Bernabei, R.; Landi, F. Persistent Symptoms in Patients After Acute COVID-19. JAMA 2020, 324, 603–605. [Google Scholar] [CrossRef]
- Carvalho-Schneider, C.; Laurent, E.; Lemaignen, A.; Beaufils, E.; Bourbao-Tournois, C.; Laribi, S.; Flament, T.; Ferreira-Maldent, N.; Bruyère, F.; Stefic, K.; et al. Follow-up of adults with noncritical COVID-19 two months after symptom onset. Clin. Microbiol. Infect. 2021, 27, 258–263. [Google Scholar] [CrossRef]
- Puntmann, V.O.; Carerj, M.L.; Wieters, I.; Fahim, M.; Arendt, C.; Hoffmann, J.; Shchendrygina, A.; Escher, F.; Vasa-Nicotera, M.; Zeiher, A.M.; et al. Outcomes of Cardiovascular Magnetic Resonance Imaging in Patients Recently Recovered From Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 1265. [Google Scholar] [CrossRef]
- Garrigues, E.; Janvier, P.; Kherabi, Y.; Le Bot, A.; Hamon, A.; Gouze, H.; Doucet, L.; Berkani, S.; Oliosi, E.; Mallart, E.; et al. Post-discharge persistent symptoms and health-related quality of life after hospitalization for COVID-19. J. Infect. 2020, 81, e4–e6. [Google Scholar] [CrossRef]
- Taquet, M.; Luciano, S.; Geddes, J.R.; Harrison, P.J. Bidirectional associations between COVID-19 and psychiatric disorder: Retrospective cohort studies of 62 354 COVID-19 cases in the USA. Lancet Psychiatry 2021, 8, 130–140. [Google Scholar] [CrossRef]
- Nordvig, A.S.; Fong, K.T.; Willey, J.Z.; Thakur, K.T.; Boehme, A.K.; Vargas, W.S.; Smith, C.J.; Elkind, M.S. Potential Neurologic Manifestations of COVID-19. Neurol. Clin. Pr. 2021, 11, e135–e146. [Google Scholar] [CrossRef]
- Heneka, M.T.; Golenbock, D.; Latz, E.; Morgan, D.; Brown, R. Immediate and long-term consequences of COVID-19 infections for the development of neurological disease. Alzheimer’s Res. Ther. 2020, 12, 69. [Google Scholar] [CrossRef]
- Kaseda, E.T.; Levine, A.J. Post-traumatic stress disorder: A differential diagnostic consideration for COVID-19 survivors. Clin. Neuropsychol. 2020, 34, 1498–1514. [Google Scholar] [CrossRef]
- Ritchie, K.; Chan, D.; Watermeyer, T. The cognitive consequences of the COVID-19 epidemic: Collateral damage? Brain Commun. 2020, 2, fcaa069. [Google Scholar] [CrossRef] [PubMed]
- Patell, R.; Bogue, T.; Koshy, A.; Bindal, P.; Merrill, M.; Aird, W.C.; Bauer, K.A.; Zwicker, J.I. Postdischarge thrombosis and hemorrhage in patients with COVID-19. Blood 2020, 136, 1342–1346. [Google Scholar] [CrossRef]
- Weng, J.; Li, Y.; Li, J.; Shen, L.; Zhu, L.; Liang, Y.; Lin, X.; Jiao, N.; Cheng, S.; Huang, Y.; et al. Gastrointestinal sequelae 90 days after discharge for COVID-19. Lancet Gastroenterol. Hepatol. 2021, 6, 344–346. [Google Scholar] [CrossRef]
- Rubino, F.; Amiel, S.A.; Zimmet, P.; Alberti, G.; Bornstein, S.; Eckel, R.H.; Mingrone, G.; Boehm, B.; Cooper, M.E.; Chai, Z.; et al. New-Onset Diabetes in Covid-19. N. Engl. J. Med. 2020, 383, 789–790. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J.S.; King, K.L.; Robbins-Juarez, S.Y.; Khairallah, P.; Toma, K.; Verduzco, H.A.; Daniel, E.; Douglas, D.; Moses, A.A.; Peleg, Y.; et al. High rate of renal recovery in survivors of COVID-19 associated acute renal failure requiring renal replacement therapy. PLoS ONE 2020, 15, e0244131. [Google Scholar] [CrossRef] [PubMed]
- Phetsouphanh, C.; Darley, D.R.; Wilson, D.B.; Howe, A.; Munier, C.M.L.; Patel, S.K.; Juno, J.A.; Burrell, L.M.; Kent, S.J.; Dore, G.J.; et al. Immunological dysfunction persists for 8 months following initial mild-to-moderate SARS-CoV-2 infection. Nat. Immunol. 2022, 23, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Schultheiß, C.; Willscher, E.; Paschold, L.; Gottschick, C.; Klee, B.; Henkes, S.S.; Bosurgi, L.; Dutzmann, J.; Sedding, D.; Frese, T.; et al. The IL-1β, IL-6, and TNF cytokine triad is associated with post-acute sequelae of COVID-19. Cell Rep. Med. 2022, 3, 100663. [Google Scholar] [CrossRef]
- Luyt, C.-E.; Combes, A.; Becquemin, M.-H.; Beigelman-Aubry, C.; Hatem, S.; Brun, A.-L.; Zraik, N.; Carrat, F.; Grenier, P.A.; Richard, J.-C.M.; et al. Long-term Outcomes of Pandemic 2009 Influenza A(H1N1)-Associated Severe ARDS. Chest 2012, 142, 583–592. [Google Scholar] [CrossRef]
- Melms, J.C.; Biermann, J.; Huang, H.; Wang, Y.; Nair, A.; Tagore, S.; Katsyv, I.; Rendeiro, A.F.; Amin, A.D.; Schapiro, D.; et al. A molecular single-cell lung atlas of lethal COVID-19. Nature 2021, 595, 114–119. [Google Scholar] [CrossRef]
- Schneider, C.; Nobs, S.P.; Kurrer, M.O.; Rehrauer, H.; Thiele, C.; Kopf, M. Induction of the nuclear receptor PPAR-γ by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat. Immunol. 2014, 15, 1026–1037. [Google Scholar] [CrossRef]
- Wang, P.; Nair, M.S.; Liu, L.; Iketani, S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.D.; et al. Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1. Nature 2021, 593, 130–135. [Google Scholar] [CrossRef]
- Sudre, C.H.; Murray, B.; Varsavsky, T.; Graham, M.S.; Penfold, R.S.; Bowyer, R.C.; Pujol, J.C.; Klaser, K.; Antonelli, M.; Canas, L.S.; et al. Attributes and predictors of long COVID. Nat. Med. 2021, 27, 626–631. [Google Scholar] [CrossRef]
- Arnold, D.T.; Hamilton, F.W.; Milne, A.; Morley, A.J.; Viner, J.; Attwood, M.; Noel, A.; Gunning, S.; Hatrick, J.; Hamilton, S.; et al. Patient outcomes after hospitalisation with COVID-19 and implications for follow-up: Results from a prospective UK cohort. Thorax 2021, 76, 399–401. [Google Scholar] [CrossRef]
- Halpin, S.J.; McIvor, C.; Whyatt, G.; Adams, A.; Harvey, O.; McLean, L.; Walshaw, C.; Kemp, S.; Corrado, J.; Singh, R.; et al. Postdischarge symptoms and rehabilitation needs in survivors of COVID-19 infection: A cross-sectional evaluation. J. Med. Virol. 2021, 93, 1013–1022. [Google Scholar] [CrossRef]
- Thompson, E.J.; Williams, D.M.; Walker, A.J.; Mitchell, R.E.; Niedzwiedz, C.L.; Yang, T.C.; Huggins, C.F.; Kwong, A.S.; Silverwood, R.J.; Di Gessa, G.; et al. Risk factors for ongoing symptomatic COVID-19 and post-COVID-19 syndrome: Analyses of 10 lon-gitudinal studies and electronic health records in the UK. medRxiv 2021. [Google Scholar] [CrossRef]
- Strain, W.D.; Sherwood, O.; Banerjee, A.; Van der Togt, V.; Hishmeh, L.; Rossman, J. The Impact of COVID Vaccination on Symptoms of Long COVID: An International Survey of People with Lived Experience of Long COVID. Vaccines 2021, 10, 652. [Google Scholar] [CrossRef]
- Kuodi, P.; Gorelik, Y.; Zayyad, H.; Wertheim, O.; Wiegler, K.B.; Jabal, K.A.; Dror, A.A.; Nazzal, S.; Glikman, D.; Edelstein, M. Association between vaccination status and reported incidence of post-acute COVID-19 symptoms in Israel: A cross-sectional study of patients tested between March 2020 and November 2021. medRxiv 2022. [Google Scholar] [CrossRef]
- Rahman, M.; Imam, H.; Nahar, N.; Chowdhury, F.U.H. Third dose vaccine With BNT162b2 and its response on Long COVID after Breakthrough infections. medRxiv 2021. [Google Scholar] [CrossRef]
- Al-Aly, Z.; Bowe, B.; Xie, Y. Long COVID after breakthrough SARS-CoV-2 infection. Nat. Med. 2022, 1–7. [Google Scholar] [CrossRef]
- Yasuhara, J.; Kuno, T.; Takagi, H.; Sumitomo, N. Clinical characteristics of COVID-19 in children: A systematic review. Pediatr. Pulmonol. 2020, 55, 2565–2575. [Google Scholar] [CrossRef] [PubMed]
- Castagnoli, R.; Votto, M.; Licari, A.; Brambilla, I.; Bruno, R.; Perlini, S.; Rovida, F.; Baldanti, F.; Marseglia, G.L. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Infection in Children and Adolescents. JAMA Pediatr. 2020, 174, 882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierce, C.A.; Sy, S.; Galen, B.; Goldstein, D.Y.; Orner, E.; Keller, M.J.; Herold, K.C.; Herold, B.C. Natural mucosal barriers and COVID-19 in children. JCI Insight 2021, 6, e148694. [Google Scholar] [CrossRef]
- Loske, J.; Röhmel, J.; Lukassen, S.; Stricker, S.; Magalhães, V.G.; Liebig, J.; Chua, R.L.; Thürmann, L.; Messingschlager, M.; Seegebarth, A.; et al. Pre-activated antiviral innate immunity in the upper airways controls early SARS-CoV-2 infection in children. Nat. Biotechnol. 2022, 40, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Worlock, K.B.; Huang, N.; Lindeboom, R.G.H.; Butler, C.R.; Kumasaka, N.; Conde, C.D.; Mamanova, L.; Bolt, L.; Richardson, L.; et al. Local and systemic responses to SARS-CoV-2 infection in children and adults. Nature 2022, 602, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Riphagen, S.; Gomez, X.; Gonzalez-Martinez, C.; Wilkinson, N.; Theocharis, P. Hyperinflammatory shock in children during COVID-19 pandemic. Lancet 2020, 395, 1607–1608. [Google Scholar] [CrossRef]
- Viner, R.M.; Whittaker, E. Kawasaki-like disease: Emerging complication during the COVID-19 pandemic. Lancet 2020, 395, 1741–1743. [Google Scholar] [CrossRef]
- Ahmed, M.; Advani, S.; Moreira, A.; Zoretic, S.; Martinez, J.; Chorath, K.; Acosta, S.; Naqvi, R.; Burmeister-Morton, F.; Burmeister, F.; et al. Multisystem inflammatory syndrome in children: A systematic review. EClinicalMedicine 2020, 26, 100527. [Google Scholar] [CrossRef]
- Hoste, L.; Van Paemel, R.; Haerynck, F. Multisystem inflammatory syndrome in children related to COVID-19: A systematic review. Eur. J. Pediatr. 2021, 180, 2019–2034. [Google Scholar] [CrossRef]
- Nakamura, A.; Ikeda, K.; Hamaoka, K. Aetiological Significance of Infectious Stimuli in Kawasaki Disease. Front. Pediatr. 2019, 7, 244. [Google Scholar] [CrossRef]
- Banday, A.Z.; Arul, A.; Vignesh, P.; Singh, M.P.; Goyal, K.; Singh, S. Kawasaki disease and influenza—New lessons from old associations. Clin. Rheumatol. 2021, 40, 2991–2999. [Google Scholar] [CrossRef]
- Gruber, C.N.; Patel, R.S.; Trachtman, R.; Lepow, L.; Amanat, F.; Krammer, F.; Wilson, K.M.; Onel, K.; Geanon, D.; Tuballes, K.; et al. Mapping Systemic Inflammation and Antibody Responses in Multisystem Inflammatory Syndrome in Children (MIS-C). Cell 2020, 183, 982–995.e14. [Google Scholar] [CrossRef]
- McArdle, A.J.; Vito, O.; Patel, H.; Seaby, E.G.; Shah, P.; Wilson, C.; Broderick, C.; Nijman, R.; Tremoulet, A.H.; Munblit, D.; et al. Treatment of Multisystem Inflammatory Syndrome in Children. New Engl. J. Med. 2021, 385, 11–22. [Google Scholar] [CrossRef]
- Stephenson, T.; Pereira, S.M.P.; Shafran, R.; de Stavola, B.L.; Rojas, N.; McOwat, K.; Simmons, R.; Zavala, M.; O’Mahoney, L.; Chalder, T.; et al. Physical and mental health 3 months after SARS-CoV-2 infection (long COVID) among adolescents in England (CLoCk): A national matched cohort study. Lancet Child Adolesc. Health 2022, 6, 230–239. [Google Scholar] [CrossRef]
- Molteni, E.; Sudre, C.H.; Canas, L.S.; Bhopal, S.S.; Hughes, R.C.; Antonelli, M.; Murray, B.; Kläser, K.; Kerfoot, E.; Chen, L.; et al. Illness duration and symptom profile in symptomatic UK school-aged children tested for SARS-CoV-2. Lancet Child Adolesc. Health 2021, 5, 708–718. [Google Scholar] [CrossRef]
- Schreiber, A.; Viemann, D.; Schöning, J.; Schloer, S.; Zambrano, A.M.; Brunotte, L.; Faist, A.; Schöfbänker, M.; Hrincius, E.; Hoffmann, H.; et al. The MEK1/2-inhibitor ATR-002 efficiently blocks SARS-CoV-2 propagation and alleviates pro-inflammatory cytokine/chemokine responses. Cell. Mol. Life Sci. 2022, 79, 65. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Günl, F.; Mecate-Zambrano, A.; Rehländer, S.; Hinse, S.; Ludwig, S.; Brunotte, L. Shooting at a Moving Target—Effectiveness and Emerging Challenges for SARS-CoV-2 Vaccine Development. Vaccines 2021, 9, 1052. [Google Scholar] [CrossRef]
- Alwan, N.A. The teachings of Long COVID. Commun. Med. 2021, 1, 15. [Google Scholar] [CrossRef]
- Alwan, N.A. Lessons from Long COVID: Working with patients to design better research. Nat. Rev. Immunol. 2022, 22, 201–202. [Google Scholar] [CrossRef]

{kind=link}
| COVID-19 | HPAIV (H5N1, H7N9 etc.) | |
|---|---|---|
| Human-to-human transmission | Yes, very efficient via aerosols Transmission before symptoms onset possible | No (only rare reports) Until today, persistent incompatibility to the human receptor |
| Cell entry | Primary target cell: Type II pneumocytes, ciliated cells Human receptor: ACE2 Spike processing protease: TMPRSS2, Catepsin L | Primary target cell: Type II pneumocytes Human receptor: α-2,6-Sialic acid HA processing protease: TMPRSS2, Furin |
| Clinical manifestations | Anosmia, fatigue, cough, dyspnea, sore throat, diarrhea, nausea, pneumonia, ARDS, organ failure | Hospitalized cases: Pneumonia, ARDS, organ failure, leukopenia, lymphopenia, decreased platelets, encephalitis, septic shock |
| Disease characteristics in severe cases | Target organ: URT, lungs Organ tropism: lung, heart, kidneys, brain, gut Immune response: excessive/dysregulated cytokine levels | Target organ: URT, lungs Organ tropism: lungs, brain, heart, kidneys Immune response: excessive/dysregulated cytokine levels |
| Biomarkers and laboratory parameters for severe disease | High cytokine levels of IL-6, IL-1β, IL-2, IL-8, IL-17, G-CSF, GMCSF, IP-10, MCP-1, TNF-α Increased levels of CRP, Procalcitonin, LDH, and D-Dimers High ferritin/transferrin ratio Glycocalyx damage | High cytokine levels of IL-6, IL-8, TNF-α, CXCL9, IP-10, MCP-1 Increased levels of CRP, LDH, and D-Dimers High creatinine and aminotransferases |
| Involved immune receptors | MDA5, TLR1/2/4/5/8/9 | RIG-I, TLR3/7, PKR |
| Monoclonal antibodies approved by FDA/EMA | Sotrovimab, Bebtelovimab, Tocilizumab | |
| Direct-acting anti-viral drugs (DAA), approved by FDA/EMA (incl. emergency use) | Nucleoside analogs: Remdesivir, Molnupiravir Protease inhibitor: Paxlovid | NA inhibitor: Oseltamivir Endonuclease inhibitor: Baloxavir |
| Anti-inflammatory treatments, approved by FDA/EMA (for emergency use) | JAK1/2 inhibitor: Baricitinib | |
| Long-term symptoms | Long COVID: ‘Brain fog‘, restricted pulmonary function, cardiac symptoms, myocardial inflammation, neurological and neuropsychiatric symptoms, venous thromboembolism, gastrointestinal symptoms, new-onset diabetes, diabetic ketoacidosis, acute kidney injury, hair loss | Chronic fatigue syndrome, myocarditis, encephalopathy or encephalitis, restricted pulmonary function, multi-organ dysfunction |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faist, A.; Janowski, J.; Kumar, S.; Hinse, S.; Çalışkan, D.M.; Lange, J.; Ludwig, S.; Brunotte, L. Virus Infection and Systemic Inflammation: Lessons Learnt from COVID-19 and Beyond. Cells 2022, 11, 2198. https://doi.org/10.3390/cells11142198
Faist A, Janowski J, Kumar S, Hinse S, Çalışkan DM, Lange J, Ludwig S, Brunotte L. Virus Infection and Systemic Inflammation: Lessons Learnt from COVID-19 and Beyond. Cells. 2022; 11(14):2198. https://doi.org/10.3390/cells11142198
Chicago/Turabian StyleFaist, Aileen, Josua Janowski, Sriram Kumar, Saskia Hinse, Duygu Merve Çalışkan, Julius Lange, Stephan Ludwig, and Linda Brunotte. 2022. "Virus Infection and Systemic Inflammation: Lessons Learnt from COVID-19 and Beyond" Cells 11, no. 14: 2198. https://doi.org/10.3390/cells11142198