
Cancer-Associated Fibroblasts and Tumor Cells in Pancreatic Cancer Microenvironment and Metastasis: Paracrine Regulators, Reciprocation and Exosomes

Abstract
:Simple Summary
Abstract
1. Introduction
2. Growth Factors Act as Paracrine Signals between Cancer-Associated Fibroblasts and Pancreatic Cancer Cells
3. Sonic Hedgehog and Insulin Signaling as Paracrine and Paracrine-Reciprocal Signals between Cancer-Associated Fibroblasts and Pancreatic Cancer Cells
4. Cancer-Associated Fibroblast Subtypes and Paracrine Factors
5. Chemokines as Paracrine and Paracrine-Reciprocal Factors Secreted by Cancer-Associated Fibroblasts and Tumor Cells
6. Exosomes and Pro-Metastatic Niche in Pancreatic Cancer
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921, Erratum in: Cancer Res. 2014, 74, 4006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, M.; Mihaljevic, A.L.; Probst, P.; Heckler, M.; Klaiber, U.; Heger, U.; Büchler, M.W.; Hackert, T. Meta-analysis of recurrence pattern after resection for pancreatic cancer. Br. J. Surg. 2019, 106, 1590–1601. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Sunami, Y.; Häußler, J.; Kleeff, J. Cellular Heterogeneity of Pancreatic Stellate Cells, Mesenchymal Stem Cells, and Cancer-Associated Fibroblasts in Pancreatic Cancer. Cancers 2020, 12, 3770. [Google Scholar] [CrossRef] [PubMed]
- Wortzel, I.; Dror, S.; Kenific, C.M.; Lyden, D. Exosome-Mediated Metastasis: Communication from a Distance. Dev. Cell. 2019, 49, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Erkan, M.; Adler, G.; Apte, M.V.; Bachem, M.G.; Buchholz, M.; Detlefsen, S.; Esposito, I.; Friess, H.; Gress, T.M.; Habisch, H.J.; et al. StellaTUM: Current consensus and discussion on pancreatic stellate cell research. Gut 2012, 61, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Zhang, B.; Hu, Q.; Qin, Y.; Xu, W.; Liu, W.; Yu, X.; Xu, J. The impact of cancer-associated fibroblasts on major hallmarks of pancreatic cancer. Theranostics 2018, 8, 5072–5087. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; Tijeras-Raballand, A.; Ragulan, C.; Cros, J.; Patil, Y.; Martinet, M.; Erkan, M.; Kleeff, J.; Wilson, J.; Apte, M.; et al. Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J. Pathol. 2019, 248, 51–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biffi, G.; Tuveson, D.A. Diversity and Biology of Cancer-Associated Fibroblasts. Physiol. Rev. 2021, 101, 147–176. [Google Scholar] [CrossRef] [PubMed]
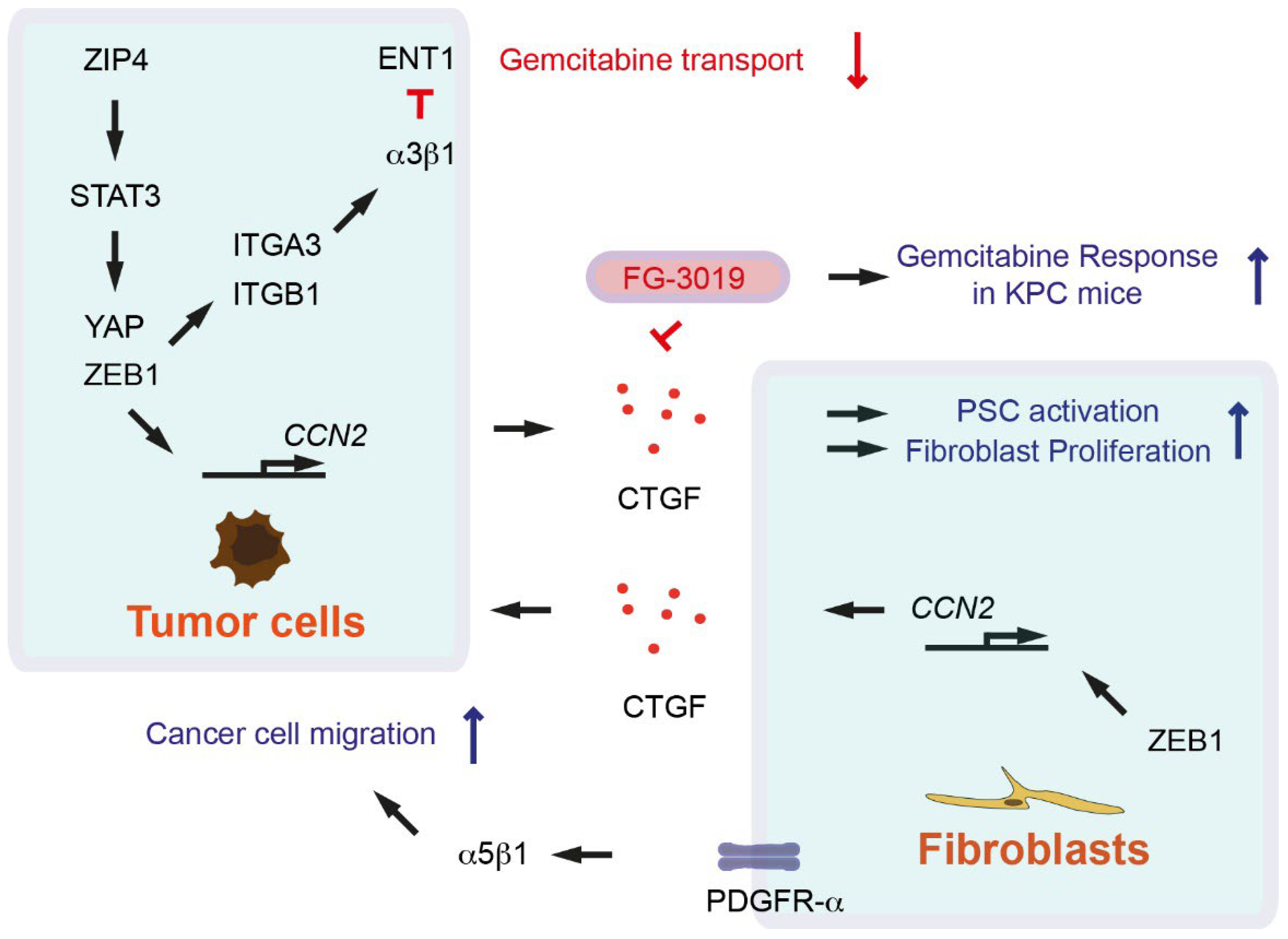
- Neesse, A.; Frese, K.K.; Bapiro, T.E.; Nakagawa, T.; Sternlicht, M.D.; Seeley, T.W.; Pilarsky, C.; Jodrell, D.I.; Spong, S.M.; Tuveson, D.A. CTGF antagonism with mAb FG-3019 enhances chemotherapy response without increasing drug delivery in murine ductal pancreas cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 12325–12330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, T.; Kodama, T.; Sato, K.; Murai, K.; Yoshioka, T.; Shigekawa, M.; Yamada, R.; Hikita, H.; Sakamori, R.; Akita, H.; et al. Dysregulation of PI3K and Hippo signaling pathways synergistically induces chronic pancreatitis via CTGF upregulation. J. Clin. Invest. 2021, 131, e143414. [Google Scholar] [CrossRef] [PubMed]
- Lobe, C.; Vallette, M.; Arbelaiz, A.; Gonzalez-Sanchez, E.; Izquierdo, L.; Pellat, A.; Guedj, N.; Louis, C.; Paradis, V.; Banales, J.M.; et al. ZEB1 promotes cholangiocarcinoma progression through tumor dedifferentiation and tumor-stroma paracrine signaling. Hepatology 2021, 74, 3194–3212. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, W.; Mossmann, D.; Kleemann, J.; Mock, K.; Meisinger, C.; Brummer, T.; Herr, R.; Brabletz, S.; Stemmler, M.P.; Brabletz, T. ZEB1 turns into a transcriptional activator by interacting with YAP1 in aggressive cancer types. Nat. Commun. 2016, 7, 10498. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, Y.; Yang, J.; Cui, X.; Zhou, Z.; Zhan, H.; Ding, K.; Tian, X.; Yang, Z.; Fung, K.A.; et al. ZIP4 Increases Expression of Transcription Factor ZEB1 to Promote Integrin α3β1 Signaling and Inhibit Expression of the Gemcitabine Transporter ENT1 in Pancreatic Cancer Cells. Gastroenterology 2020, 158, 679–692.e1. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhang, Y.; Yang, J.; Zhan, H.; Zhou, Z.; Jiang, Y.; Shi, X.; Fan, X.; Zhang, J.; Luo, W.; et al. Zinc-Dependent Regulation of ZEB1 and YAP1 Coactivation Promotes Epithelial-Mesenchymal Transition Plasticity and Metastasis in Pancreatic Cancer. Gastroenterology 2021, 160, 1771–1783.e1. [Google Scholar] [CrossRef] [PubMed]
- Charrier, A.; Brigstock, D.R. Regulation of pancreatic function by connective tissue growth factor (CTGF, CCN2). Cytokine Growth Factor Rev. 2013, 24, 59–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tape, C.J.; Ling, S.; Dimitriadi, M.; McMahon, K.M.; Worboys, J.D.; Leong, H.S.; Norrie, I.C.; Miller, C.J.; Poulogiannis, G.; Lauffenburger, D.A.; et al. Oncogenic KRAS Regulates Tumor Cell Signaling via Stromal Reciprocation. Cell 2016, 165, 910–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
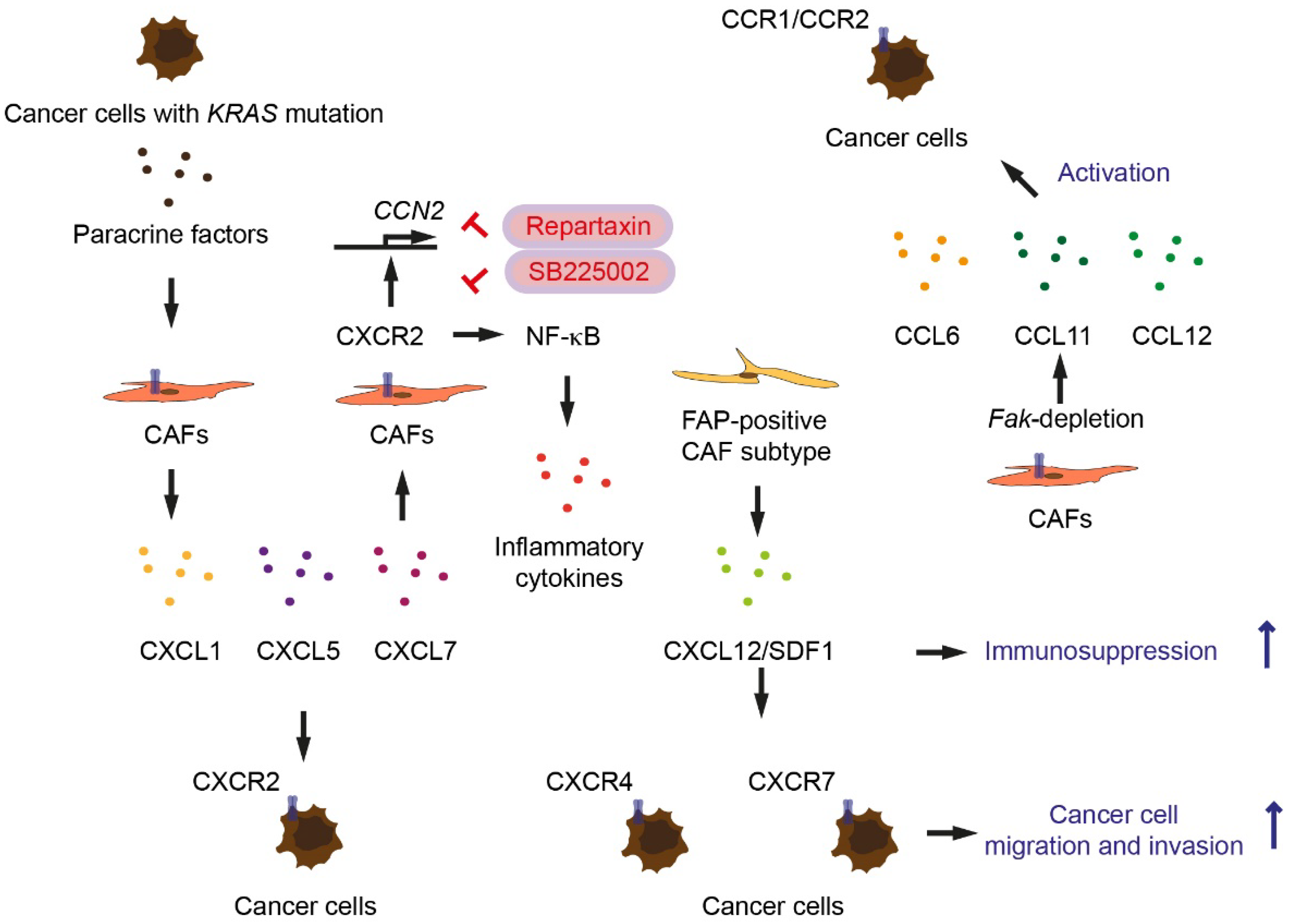
- Awaji, M.; Saxena, S.; Wu, L.; Prajapati, D.R.; Purohit, A.; Varney, M.L.; Kumar, S.; Rachagani, S.; Ly, Q.P.; Jain, M.; et al. CXCR2 signaling promotes secretory cancer-associated fibroblasts in pancreatic ductal adenocarcinoma. FASEB J. 2020, 34, 9405–9418. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Kim, J.; Yang, S.; Wang, H.; Wu, C.J.; Sugimoto, H.; LeBleu, V.S.; Kalluri, R. Type I collagen deletion in αSMA+ myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell 2021, 39, 548–565.e6. [Google Scholar] [CrossRef] [PubMed]
- Ijichi, H.; Chytil, A.; Gorska, A.E.; Aakre, M.E.; Bierie, B.; Tada, M.; Mohri, D.; Miyabayashi, K.; Asaoka, Y.; Maeda, S.; et al. Inhibiting Cxcr2 disrupts tumor-stromal interactions and improves survival in a mouse model of pancreatic ductal adenocarcinoma. J. Clin. Invest. 2011, 121, 4106–4117. [Google Scholar] [CrossRef] [PubMed]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [Green Version]
- Demircioglu, F.; Wang, J.; Candido, J.; Costa, A.S.H.; Casado, P.; de Luxan Delgado, B.; Reynolds, L.E.; Gomez-Escudero, J.; Newport, E.; Rajeeve, V.; et al. Cancer associated fibroblast FAK regulates malignant cell metabolism. Nat. Commun. 2020, 11, 1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardito, C.M.; Grüner, B.M.; Takeuchi, K.K.; Lubeseder-Martellato, C.; Teichmann, N.; Mazur, P.K.; Delgiorno, K.E.; Carpenter, E.S.; Halbrook, C.J.; Hall, J.C.; et al. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell 2012, 22, 304–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navas, C.; Hernández-Porras, I.; Schuhmacher, A.J.; Sibilia, M.; Guerra, C.; Barbacid, M. EGF receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell 2012, 22, 318–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahajan, U.M.; Li, Q.; Alnatsha, A.; Maas, J.; Orth, M.; Maier, S.H.; Peterhansl, J.; Regel, I.; Sendler, M.; Wagh, P.R.; et al. Tumor-Specific Delivery of 5-Fluorouracil-Incorporated Epidermal Growth Factor Receptor-Targeted Aptamers as an Efficient Treatment in Pancreatic Ductal Adenocarcinoma Models. Gastroenterology 2021, 161, 996–1010. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yuan, S.; Norgard, R.J.; Yan, F.; Sun, Y.H.; Kim, I.K.; Merrell, A.J.; Sela, Y.; Jiang, Y.; Bhanu, N.V.; et al. Epigenetic and Transcriptional Control of the Epidermal Growth Factor Receptor Regulates the Tumor Immune Microenvironment in Pancreatic Cancer. Cancer Discov. 2021, 11, 736–753. [Google Scholar] [CrossRef] [PubMed]
- Weissmueller, S.; Manchado, E.; Saborowski, M.; Morris JP 4th Wagenblast, E.; Davis, C.A.; Moon, S.H.; Pfister, N.T.; Tschaharganeh, D.F.; Kitzing, T.; Aust, D.; et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor β signaling. Cell 2014, 157, 382–394. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, S.; Furukawa, S.; Hashimoto, A.; Tsutaho, A.; Fukao, A.; Sakamura, Y.; Parajuli, G.; Onodera, Y.; Otsuka, Y.; Handa, H.; et al. ARF6 and AMAP1 are major targets of KRAS and TP53 mutations to promote invasion, PD-L1 dynamics, and immune evasion of pancreatic cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 17450–17459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurahara, H.; Maemura, K.; Mataki, Y.; Sakoda, M.; Shinchi, H.; Natsugoe, S. Impact of p53 and PDGFR-β Expression on Metastasis and Prognosis of Patients with Pancreatic Cancer. World J. Surg. 2016, 40, 1977–1984. [Google Scholar] [CrossRef] [PubMed]
- Yuzawa, S.; Kano, M.R.; Einama, T.; Nishihara, H. PDGFRβ expression in tumor stroma of pancreatic adenocarcinoma as a reliable prognostic marker. Med. Oncol. 2012, 29, 2824–2830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdogan, B.; Ao, M.; White, L.M.; Means, A.L.; Brewer, B.M.; Yang, L.; Washington, M.K.; Shi, C.; Franco, O.E.; Weaver, A.M.; et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J. Cell Biol. 2017, 216, 3799–3816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
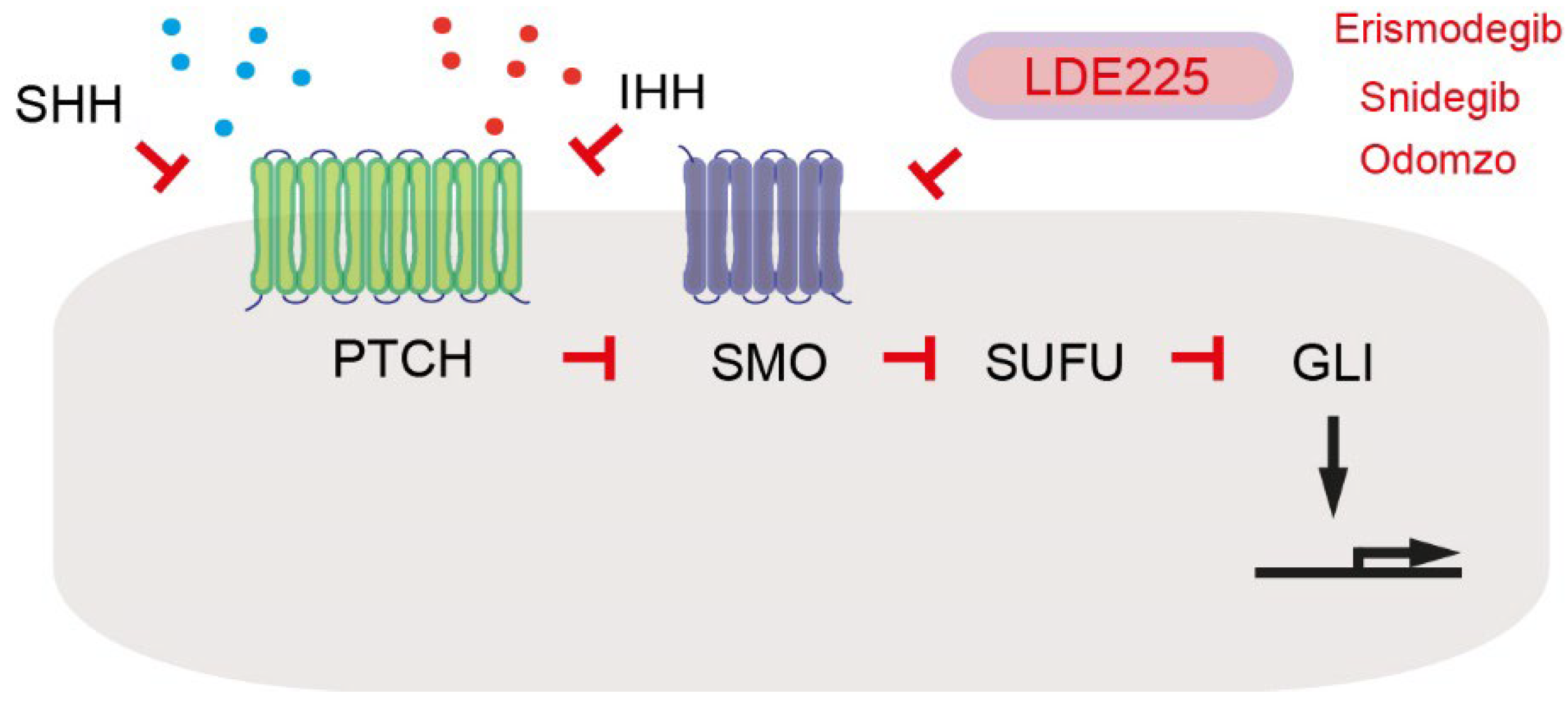
- Sigafoos, A.N.; Paradise, B.D.; Fernandez-Zapico, M.E. Hedgehog/GLI Signaling Pathway: Transduction, Regulation, and Implications for Disease. Cancers 2021, 13, 3410. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Wei, Y.; Wei, X. AXL receptor tyrosine kinase as a promising anti-cancer approach: Functions, molecular mechanisms and clinical applications. Mol. Cancer 2019, 18, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steele, N.G.; Biffi, G.; Kemp, S.B.; Zhang, Y.; Drouillard, D.; Syu, L.; Hao, Y.; Oni, T.E.; Brosnan, E.; Elyada, E.; et al. Inhibition of Hedgehog Signaling Alters Fibroblast Composition in Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 2023–2037. [Google Scholar] [CrossRef] [PubMed]
- Rimkus, T.K.; Carpenter, R.L.; Qasem, S.; Chan, M.; Lo, H.W. Targeting the Sonic Hedgehog Signaling Pathway: Review of Smoothened and GLI Inhibitors. Cancers 2016, 8, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Pitarresi, J.R.; Cuitiño, M.C.; Kladney, R.D.; Woelke, S.A.; Sizemore, G.M.; Nayak, S.G.; Egriboz, O.; Schweickert, P.G.; Yu, L.; et al. Genetic ablation of Smoothened in pancreatic fibroblasts increases acinar-ductal metaplasia. Genes Dev. 2016, 30, 1943–1955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Gao, W.; Lytle, N.K.; Huang, P.; Yuan, X.; Dann, A.M.; Ridinger-Saison, M.; DelGiorno, K.E.; Antal, C.E.; Liang, G.; et al. Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature 2019, 569, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.A.; McIntosh, B.J.; Shi, Y.; Sperberg, R.A.P.; Funatogawa, C.; Labanieh, L.; Soon, E.; Wastyk, H.C.; Mehta, N.; Carter, C.; et al. An engineered ligand trap inhibits leukemia inhibitory factor as pancreatic cancer treatment strategy. Commun. Biol. 2021, 4, 452. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Bai, W.; Li, J.; Liu, J.; Zhao, K.; Ren, L. Leukemia inhibitory factor is a novel biomarker to predict lymph node and distant metastasis in pancreatic cancer. Int. J. Cancer 2021, 148, 1006–1013. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Gandler, H.I.; Tošić, I.; Ye, D.Q.; Giaccone, Z.T.; Frank, D.A. Mutant KRAS Downregulates the Receptor for Leukemia Inhibitory Factor (LIF) to Enhance a Signature of Glycolysis in Pancreatic Cancer and Lung Cancer. Mol. Cancer Res. 2021, 19, 1283–1295. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.T.; Fer, N.; Galeas, J.; Collisson, E.A.; Kim, S.E.; Sharib, J.; McCormick, F. Blockade of leukemia inhibitory factor as a therapeutic approach to KRAS driven pancreatic cancer. Nat. Commun. 2019, 10, 3055. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Ma, X.L.; Wei, Y.Q.; Wei, X.W. Potential roles and targeted therapy of the CXCLs/CXCR2 axis in cancer and inflammatory diseases. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 289–312. [Google Scholar] [CrossRef] [PubMed]
- Seifert, L.; Werba, G.; Tiwari, S.; Giao Ly, N.N.; Alothman, S.; Alqunaibit, D.; Avanzi, A.; Barilla, R.; Daley, D.; Greco, S.H.; et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature 2016, 532, 245–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aue, E.; Wirsik, N.M.; Freire Valls, A.; Shen, Y.; Radhakrishnan, P.; Hackert, T.; Schneider, M.; Schmidt, T. Metastasis-associated fibroblasts promote angiogenesis in metastasized pancreatic cancer via the CXCL8 and the CCL2 axes. Sci. Rep. 2020, 10, 5420. [Google Scholar] [CrossRef]
- Purohit, A.; Saxena, S.; Varney, M.; Prajapati, D.R.; Kozel, J.A.; Lazenby, A.; Singh, R.K. Host Cxcr2-Dependent Regulation of Pancreatic Cancer Growth, Angiogenesis, and Metastasis. Am. J. Pathol. 2021, 191, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Lesina, M.; Wörmann, S.M.; Morton, J.; Diakopoulos, K.N.; Korneeva, O.; Wimmer, M.; Einwächter, H.; Sperveslage, J.; Demir, I.E.; Kehl, T.; et al. RelA regulates CXCL1/CXCR2-dependent oncogene-induced senescence in murine Kras-driven pancreatic carcinogenesis. J. Clin. Invest. 2016, 126, 2919–2932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ene-Obong, A.; Clear, A.J.; Watt, J.; Wang, J.; Fatah, R.; Riches, J.C.; Marshall, J.F.; Chin-Aleong, J.; Chelala, C.; Gribben, J.G.; et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology 2013, 145, 1121–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, B.; Giri, B.; Modi, S.; Sethi, V.; Castro, I.; Umland, O.; Ban, Y.; Lavania, S.; Dawra, R.; Banerjee, S.; et al. NFκB in Pancreatic Stellate Cells Reduces Infiltration of Tumors by Cytotoxic T Cells and Killing of Cancer Cells, via Up-regulation of CXCL12. Gastroenterology 2018, 155, 880–891.e8. [Google Scholar] [CrossRef] [PubMed]
- Khare, T.; Bissonnette, M.; Khare, S. CXCL12-CXCR4/CXCR7 Axis in Colorectal Cancer: Therapeutic Target in Preclinical and Clinical Studies. Int. J. Mol. Sci. 2021, 22, 7371. [Google Scholar] [CrossRef] [PubMed]
- Bockorny, B.; Semenisty, V.; Macarulla, T.; Borazanci, E.; Wolpin, B.M.; Stemmer, S.M.; Golan, T.; Geva, R.; Borad, M.J.; Pedersen, K.S.; et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: The COMBAT trial. Nat. Med. 2020, 26, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Morita, T.; Kodama, Y.; Shiokawa, M.; Kuriyama, K.; Marui, S.; Kuwada, T.; Sogabe, Y.; Matsumori, T.; Kakiuchi, N.; Tomono, T.; et al. CXCR4 in Tumor Epithelial Cells Mediates Desmoplastic Reaction in Pancreatic Ductal Adenocarcinoma. Cancer Res. 2020, 80, 4058–4070. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.C.; Li, J.; Zhou, L.; Yang, J.Y.; Zhang, Z.G.; Liang, Z.Y.; Zhou, W.X.; You, L.; Zhang, T.P.; Zhao, Y.P. CXCL12-CXCR7 axis contributes to the invasive phenotype of pancreatic cancer. Oncotarget 2016, 7, 62006–62018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Hiraoka, N.; Onozato, K.; Kosuge, T.; Hirohashi, S. Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin. Cancer Res. 2006, 12, 5423–5434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Lazarus, J.; Steele, N.G.; Yan, W.; Lee, H.J.; Nwosu, Z.C.; Halbrook, C.J.; Menjivar, R.E.; Kemp, S.B.; Sirihorachai, V.R.; et al. Regulatory T-cell Depletion Alters the Tumor Microenvironment and Accelerates Pancreatic Carcinogenesis. Cancer Discov. 2020, 10, 422–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and chemokine receptors: Positioning cells for host defense and immunity. Annu. Rev. Immunol. 2014, 32, 659–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R. The biology and function of exosomes in cancer. J. Clin. Invest. 2016, 126, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Bernard, V.; Kim, D.U.; San Lucas, F.A.; Castillo, J.; Allenson, K.; Mulu, F.C.; Stephens, B.M.; Huang, J.; Semaan, A.; Guerrero, P.A.; et al. Circulating Nucleic Acids Are Associated with Outcomes of Patients with Pancreatic Cancer. Gastroenterology 2019, 156, 108–118.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, D.D.; Gercel-Taylor, C. Exosomes/microvesicles: Mediators of cancer-associated immunosuppressive microenvironments. Semin. Immunopathol. 2011, 33, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Yamamoto, H.; Harada, T.; Fumoto, K.; Osugi, Y.; Sada, R.; Maehara, N.; Hikita, H.; Mori, S.; Eguchi, H.; et al. CKAP4, a DKK1 Receptor, is a Biomarker in Exosomes Derived from Pancreatic Cancer and a Molecular Target for Therapy. Clin. Cancer Res. 2019, 25, 1936–1947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melo, S.A.; Luecke, L.B.; Kahlert, C.; Fernandez, A.F.; Gammon, S.T.; Kaye, J.; LeBleu, V.S.; Mittendorf, E.A.; Weitz, J.; Rahbari, N.; et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature 2015, 523, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, K.S.; Ciprani, D.; O’Shea, A.; Liss, A.S.; Yang, R.; Fletcher-Mercaldo, S.; Mino-Kenudson, M.; Fernández-Del Castillo, C.; Weissleder, R. Extracellular Vesicle Analysis Allows for Identification of Invasive IPMN. Gastroenterology 2021, 160, 1345–1358.e11. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Dong, Z.; Zhen, L.; Xia, G.; Huang, X.; Wang, T.; Guo, H.; Yang, B.; Xu, C.; Wu, W.; et al. Combined Exosomal GPC1, CD82, and Serum CA19-9 as Multiplex Targets: A Specific, Sensitive, and Reproducible Detection Panel for the Diagnosis of Pancreatic Cancer. Mol. Cancer Res. 2020, 18, 300–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsujii, S.; Serada, S.; Fujimoto, M.; Uemura, S.; Namikawa, T.; Nomura, T.; Murakami, I.; Hanazaki, K.; Naka, T. Glypican-1 is a novel target for stroma and tumor cell dual-targeting antibody-drug conjugates in pancreatic cancer. Mol. Cancer Ther. 2021, 20, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- Richards, K.E.; Zeleniak, A.E.; Fishel, M.L.; Wu, J.; Littlepage, L.E.; Hill, R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene 2017, 36, 1770–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Essandoh, K.; Yang, L.; Wang, X.; Huang, W.; Qin, D.; Hao, J.; Wang, Y.; Zingarelli, B.; Peng, T.; Fan, G.C. Blockade of exosome generation with GW4869 dampens the sepsis-induced inflammation and cardiac dysfunction. Biochim. Biophys. Acta 2015, 1852, 2362–2371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Döppler, H.; Braun, U.B.; Panayiotou, R.; Scotti Buzhardt, M.; Radisky, D.C.; Crawford, H.C.; Fields, A.P.; Murray, N.R.; Wang, Q.J.; et al. Protein kinase D1 drives pancreatic acinar cell reprogramming and progression to intraepithelial neoplasia. Nat. Commun. 2015, 6, 6200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armacki, M.; Polaschek, S.; Waldenmaier, M.; Morawe, M.; Ruhland, C.; Schmid, R.; Lechel, A.; Tharehalli, U.; Steup, C.; Bektas, Y.; et al. Protein Kinase D1, Reduced in Human Pancreatic Tumors, Increases Secretion of Small Extracellular Vesicles from Cancer Cells That Promote Metastasis to Lung in Mice. Gastroenterology 2020, 159, 1019–1035.e22. [Google Scholar] [CrossRef] [PubMed]
- Novo, D.; Heath, N.; Mitchell, L.; Caligiuri, G.; MacFarlane, A.; Reijmer, D.; Charlton, L.; Knight, J.; Calka, M.; McGhee, E.; et al. Mutant p53s generate pro-invasive niches by influencing exosome podocalyxin levels. Nat. Commun. 2018, 9, 5069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Factor | Source | Mode of Action | Functional Relevance | Reference |
|---|---|---|---|---|
| CTGF | CAFs, tumor cells from KPC mice | Paracrine | Act on CAFs and tumor cells | [11] |
| Pancreatic cancer cells | Paracrine | Promotes proliferation, adhesion, migration of PSCs | [17] | |
| HGF, IL-6 | CAFs, tumor cells | Paracrine | Act on CAFs and tumor cells | [13] |
| SHH | Pancreatic tumor cells | Paracrine | Induces expression of IGF1 and GAS6 in PSCs | [18] |
| IGF1 | PSCs | Paracrine- reciprocal | Activates IGF1R signaling | [18] |
| GAS6 | PSCs | Paracrine- reciprocal | Activates AXL signaling | [18] |
| IL-1α | Pancreatic tumor cells | Paracrine | Activates NF-κB signaling and expression of LIF in iCAFs | [19] |
| LIF | iCAFs | Autocrine | Activates JAK/STAT signaling and establishes a positive feedback loop by upregulating IL-1R1 | [19] |
| LIF | iCAFs | Paracrine (-reciprocal) | Activate STAT3 signaling in cancer cells | [19] |
| KRAS-driven factors (unknown) | Pancreatic cells | Paracrine | Act on CAFs inducing secretion of CXCR2 ligands and CXCR2 expression | [20] |
| CXCR2 ligands | CAFs | Autocrine | CXCR2 signaling in CAFs | [20] |
| CXCR2 ligands | Pancreatic tumor cells | Paracrine | Recruit MDSCs | [21] |
| CXCR2 ligands | Pancreatic cancer cells from Ptf1a-Cre; lox-stop-lox-KrasG12D/+; Tgfbr2lox/lox mice | Paracrine | Induce Ctgf expression in CAFs | [22] |
| CXCL12 | CAFs | Paracrine | Immunosuppression | [23] |
| CCL6, CCL11, CCL12 | Fak-depleted CAFs | Paracrine | Activate CCR1/CCR2 on cancer cells | [24] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sunami, Y.; Häußler, J.; Zourelidis, A.; Kleeff, J. Cancer-Associated Fibroblasts and Tumor Cells in Pancreatic Cancer Microenvironment and Metastasis: Paracrine Regulators, Reciprocation and Exosomes. Cancers 2022, 14, 744. https://doi.org/10.3390/cancers14030744
Sunami Y, Häußler J, Zourelidis A, Kleeff J. Cancer-Associated Fibroblasts and Tumor Cells in Pancreatic Cancer Microenvironment and Metastasis: Paracrine Regulators, Reciprocation and Exosomes. Cancers. 2022; 14(3):744. https://doi.org/10.3390/cancers14030744
Chicago/Turabian StyleSunami, Yoshiaki, Johanna Häußler, Anais Zourelidis, and Jörg Kleeff. 2022. "Cancer-Associated Fibroblasts and Tumor Cells in Pancreatic Cancer Microenvironment and Metastasis: Paracrine Regulators, Reciprocation and Exosomes" Cancers 14, no. 3: 744. https://doi.org/10.3390/cancers14030744