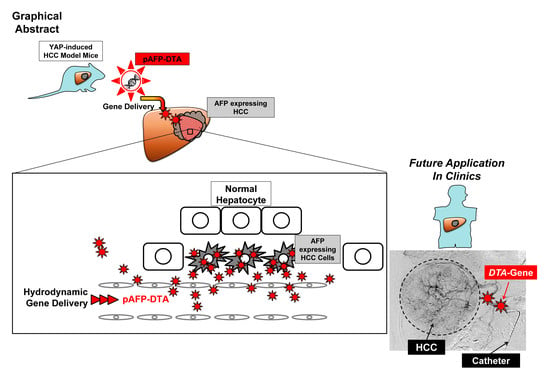
Effect of Diphtheria Toxin-Based Gene Therapy for Hepatocellular Carcinoma

 , , ,
, , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Development of DTA-Expressing Plasmid
2.2. Effect of DTA Gene Expression in Mice Liver on Protein Synthesis
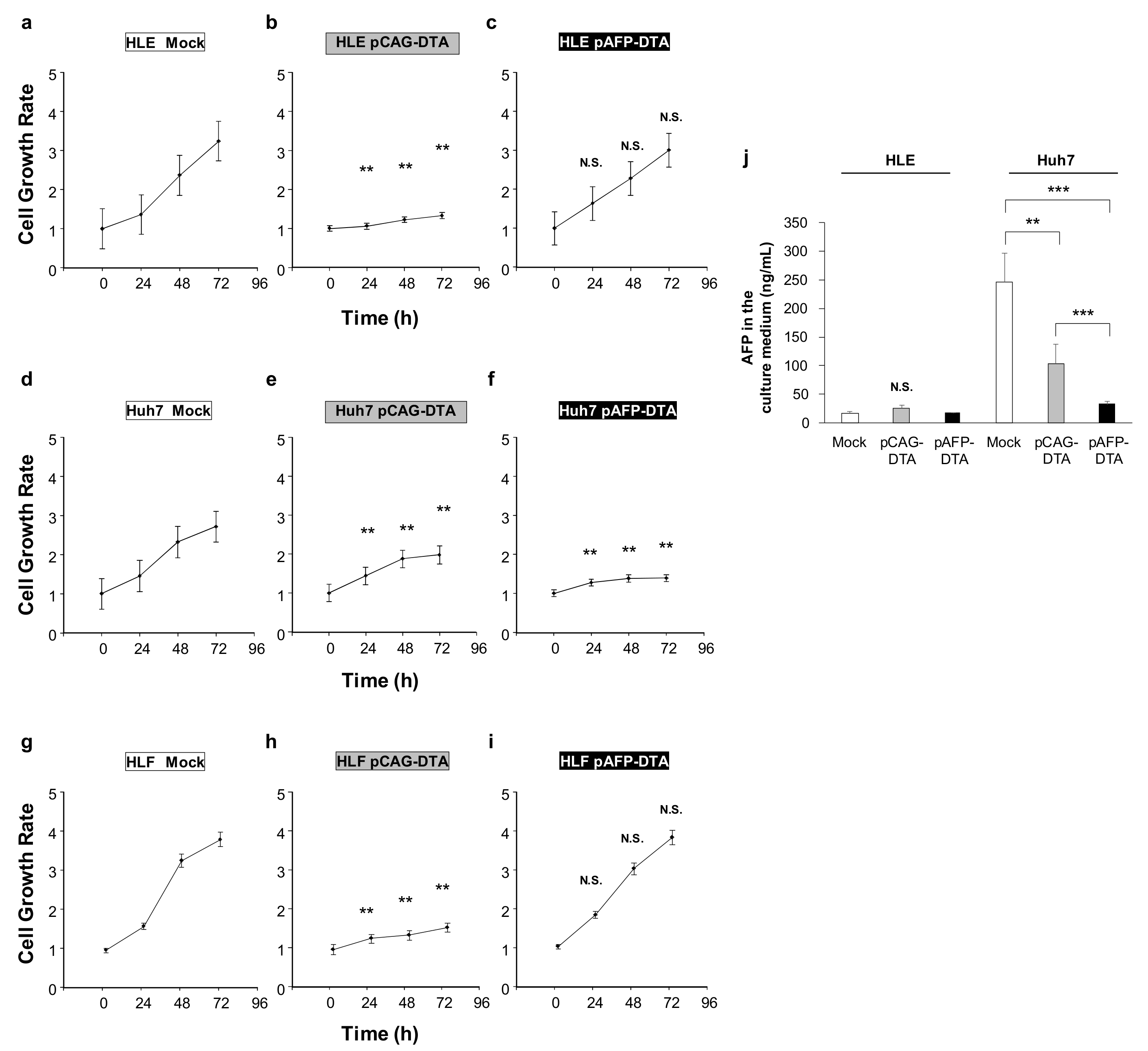
2.3. Effect of DTA Expression in HCC Cells on the Cell Growth
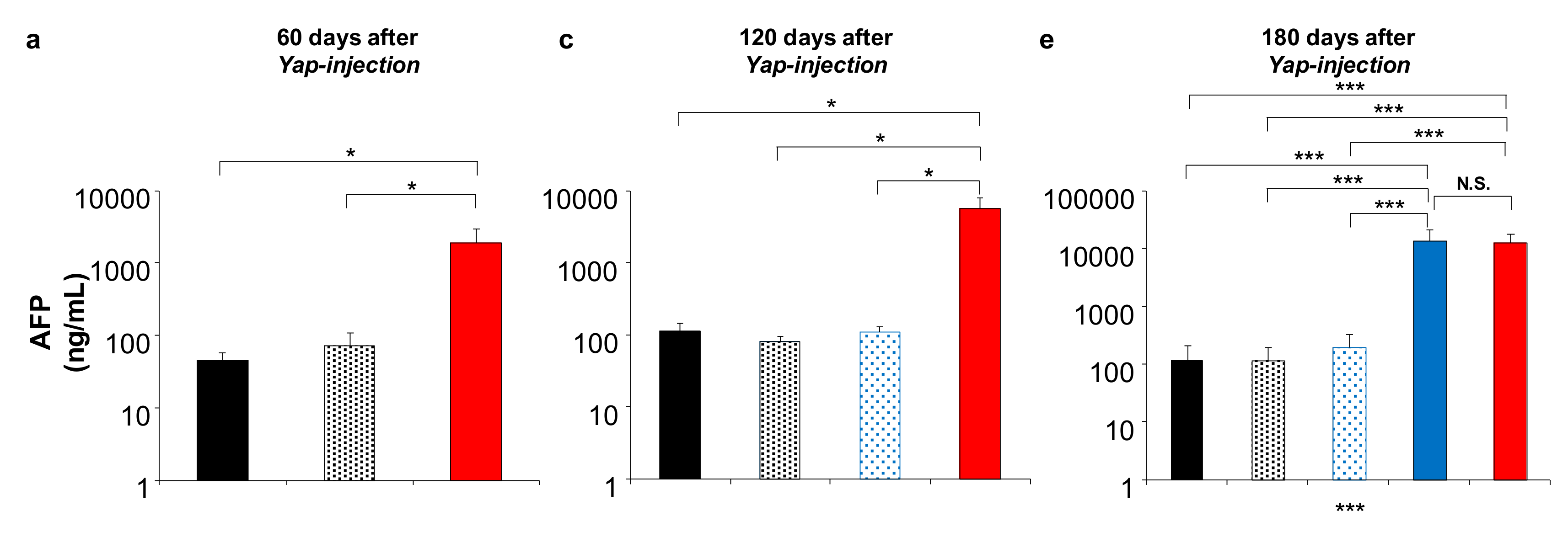
2.4. Effect of DTA on Tumor Growth In Vivo
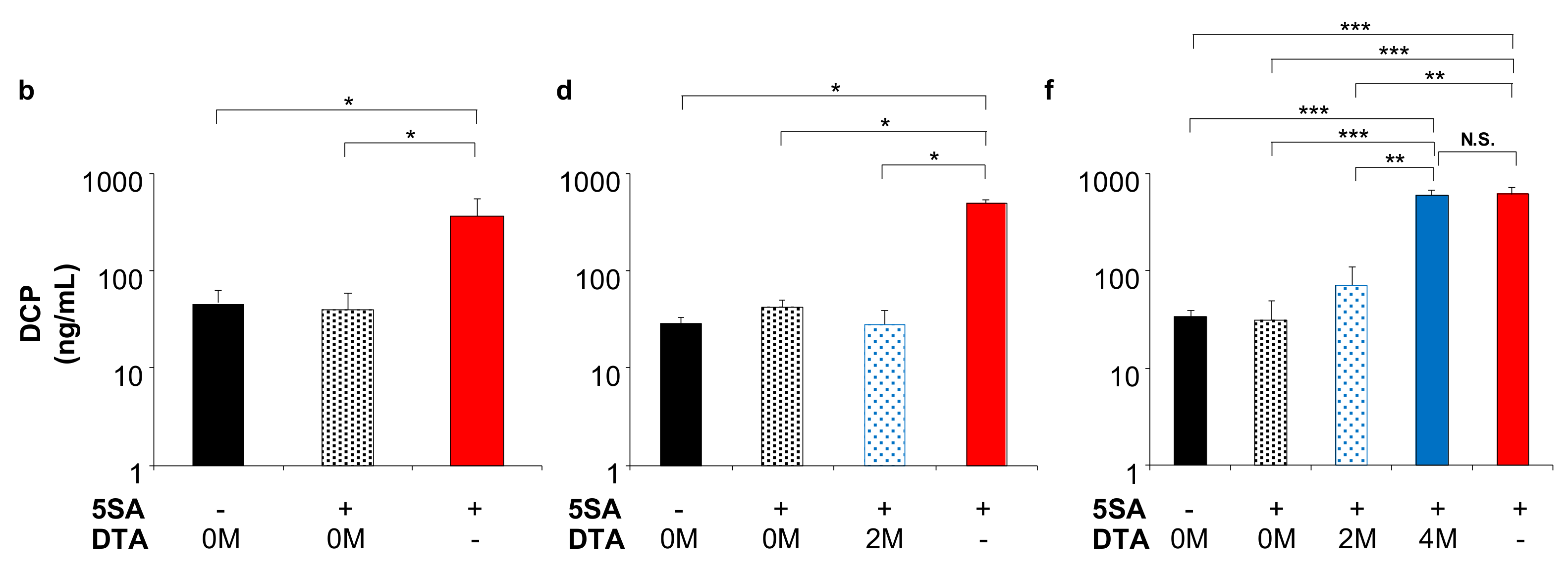
2.5. Effect of DTA on HCC Tumor Marker In Vivo
2.6. Safety of the DTA Gene Therapy with Promoter Selectivity
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Plasmids
4.3. Cells
4.4. Cell Growth Assay
4.5. Fluorescence Image
4.6. Histological Analysis
4.7. Serum Biochemical Analysis
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef]
- Bruix, J.; Reig, M.; Sherman, M. Evidence-based diagnosis, staging, and treatment of patients with hepatocellular carcinoma. Gastroenterology 2016, 150, 835–853. [Google Scholar] [CrossRef] [Green Version]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular Carcinoma. Nat. Rev. Dis. Primers. 2016, 2, 16018. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. European association for the study of the liver EASL clinical practice guidelines: management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar]
- Llovet, J.M.; Montal, R.; Villanueva, A. Randomized trials and endpoints in advanced hcc: role of pfs as a surrogate of survival. J. Hepatol. 2019, 70, 1262–1277. [Google Scholar] [CrossRef] [Green Version]
- Calderaro, J.; Ziol, M.; Paradis, V.; Zucman-Rossi, J. Molecular and histological correlations in liver cancer. J. Hepatol. 2019, 71, 616–630. [Google Scholar] [CrossRef] [Green Version]
- El Dika, I.; Khalil, D.N.; Abou-Alfa, G.K. Immune checkpoint inhibitors for hepatocellular carcinoma. Cancer 2019, 125, 3312–3319. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, S.; Zeng, S.; Shen, H. From Bench to Bed: The tumor immune microenvironment and current immunotherapeutic strategies for hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 396. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Rong, D.; Zhang, B.; Zheng, W.; Wang, X.; Chen, Z.; Tang, W. Current perspectives on the immunosuppressive tumor microenvironment in hepatocellular carcinoma: challenges and opportunities. Mol. Cancer 2019, 18, 130. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Zhang, H.; Sun, B.; Karin, M. The immunobiology of hepatocellular carcinoma in humans and mice: basic concepts and therapeutic implications. J. Hepatol. 2020, 72, 167–182. [Google Scholar] [CrossRef]
- Alaei-Mahabadi, B.; Bhadury, J.; Karlsson, J.W.; Nilsson, J.A.; Larsson, E. Global analysis of somatic structural genomic alterations and their impact on gene expression in diverse human cancers. Proc. Natl. Acad. Sci. USA 2016, 113, 13768–13773. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, A.; Furuta, M.; Totoki, Y.; Tsunoda, T.; Kato, M.; Shiraishi, Y.; Tanaka, H.; Taniguchi, H.; Kawakami, Y.; Ueno, M.; et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat. Genet. 2016, 48, 500–509. [Google Scholar] [CrossRef]
- Rao, C.V.; Asch, A.S.; Yamada, H.Y. Frequently mutated genes/pathways and genomic instability as prevention targets in liver cancer. Carcinogenesis 2017, 38, 2–11. [Google Scholar] [CrossRef] [Green Version]
- Gillet, J.P.; Andersen, J.B.; Madigan, J.P.; Varma, S.; Bagni, R.K.; Powell, K.; Burgan, W.E.; Wu, C.P.; Calcagno, A.M.; Ambudkar, S.V.; et al. A gene expression signature associated with overall survival in patients with hepatocellular carcinoma suggests a new treatment strategy. Mol. Pharmacol. 2016, 89, 263–272. [Google Scholar] [CrossRef] [Green Version]
- Kanda, T.; Moriyama, M.; Omata, M. Molecular Mechanisms Driving Progression of Liver Cirrhosis towards Hepatocellular Carcinoma in Chronic Hepatitis B and C Infections: A Review. Int. J. Mol. Sci. 2019, 20, 1358. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Shi, Q.; Zhang, H.; Yang, K.; Ke, Y.; Wang, Y.; Qiao, L. Advances in the techniques and methodologies of cancer-gene therapy. Discov. Med. 2019, 27, 45–55. [Google Scholar]
- Munoz-Garrido, P.; Andersen, J.B. Genetic optimization of liver cancer therapy: A patient-derived primary cancer cell-based model. Gastroenterology 2017, 152, 19–21. [Google Scholar] [CrossRef] [Green Version]
- Gao, Q.; Wang, Z.C.; Duan, M.; Lin, Y.H.; Zhou, X.Y.; Worthley, D.L.; Wang, X.Y.; Niu, G.; Xia, Y.; Deng, M.; et al. Cell culture system for analysis of genetic heterogeneity within hepatocellular carcinomas and response to pharmacologic agents. Gastroenterology 2017, 152, 232–242. [Google Scholar] [CrossRef] [Green Version]
- Dunbar, C.E.; High, K.A.; Joung, J.K.; Kohn, D.B.; Ozawa, K.; Sadelain, M. Gene therapy comes of age. Science 2018, 359, eaan4672. [Google Scholar] [CrossRef] [Green Version]
- Naldini, L. Gene therapy returns to centre stage. Nature 2015, 526, 351–360. [Google Scholar] [CrossRef] [PubMed]
- High, K.A.; Roncarolo, M.G. Gene Therapy. N. Engl. J. Med. 2019, 381, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Ginn, S.L.; Amaya, A.K.; Alexander, I.E.; Edelstein, M.; Abedi, M.R. Gene therapy clinical trials worldwide to 2017: An update. J. Gene Med. 2018, 20, e3015. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tai, Z.; Zhang, W.; Gao, S. Current status of gene therapy for hepatocellular carcinoma, with a focus on gene delivery approaches. Curr. Gene Ther. 2015, 15, 120–141. [Google Scholar] [CrossRef]
- Reghupaty, S.C.; Sarkar, D. Current status of gene therapy in hepatocellular carcinoma. Cancers 2019, 11, 1265. [Google Scholar] [CrossRef] [Green Version]
- Kamimura, K.; Yokoo, T.; Abe, H.; Terai, S. Gene therapy for liver cancers: current status from basic to clinics. Cancers 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 2017, 169, 1327–1341. [Google Scholar] [CrossRef]
- Lai, Y.H.; Lin, C.C.; Chen, S.H.; Tai, C.K. Tumor-specific suicide gene therapy for hepatocellular carcinoma by transcriptionally targeted retroviral replicating vectors. Gene Ther. 2015, 22, 155–162. [Google Scholar] [CrossRef]
- Chen, Z.H.; Yu, Y.P.; Zuo, Z.H.; Nelson, J.B.; Michalopoulos, G.K.; Monga, S.; Liu, S.; Tseng, G.; Luo, J.H. Targeting genomic rearrangements in tumor cells through Cas9-mediated insertion of a suicide gene. Nat. Biotechnol. 2017, 35, 543–550. [Google Scholar] [CrossRef] [Green Version]
- Sangro, B.; Mazzolini, G.; Ruiz, M.; Ruiz, J.; Quiroga, J.; Herrero, I.; Qian, C.; Benito, A.; Larrache, J.; Olagüe, C.; et al. A Phase I clinical trial of thymidine kinase-based gene therapy in advanced hepatocellular carcinoma. Cancer Gene Ther. 2010, 17, 837–843. [Google Scholar] [CrossRef]
- Düzgüneş, N. Origins of Suicide Gene Therapy. Methods Mol. Biol. 2019, 1895, 1–9. [Google Scholar]
- Lin, B.; Gao, A.; Zhang, R.; Ma, H.; Shen, H.; Hu, Q.; Zhang, H.; Zhao, M.; Lan, X.; Liu, K. Use of a novel integrase-deficient lentivirus for targeted anti-cancer therapy with survivin promoter-driven diphtheria toxin A. Medicine 2015, 94, e1301. [Google Scholar] [CrossRef]
- Shafiee, F.; Aucoin, M.G.; Jahanian-Najafabadi, A. Targeted diphtheria toxin-based therapy: A review article. Front. Microbiol. 2019, 10, 2340. [Google Scholar] [CrossRef]
- Hanna, N.; Ohana, P.; Konikoff, F.M.; Leichtmann, G.; Hubert, A.; Appelbaum, L.; Kopelman, Y.; Czerniak, A.; Hochberg, A. Phase 1/2a, Dose-escalation, safety, pharmacokinetic and preliminary efficacy study of intratumoral administration of BC-819 in patients with unresectable pancreatic cancer. Cancer Gene Ther. 2012, 19, 374–381. [Google Scholar] [CrossRef]
- Scaiewicz, V.; Sorin, V.; Fellig, Y.; Birman, T.; Mizrahi, A.; Galula, J.; Abu-Lail, R.; Shneider, T.; Ohana, P.; Buscail, L.; et al. Use of H19 gene regulatory sequences in DNA-based therapy for pancreatic cancer. J. Oncol. 2010, 2010, 178174. [Google Scholar] [CrossRef] [Green Version]
- Mizrahi, A.; Czerniak, A.; Levy, T.; Amiur, S.; Gallula, J.; Matouk, I.; Abu-lail, R.; Sorin, V.; Birman, T.; de Groot, N.; et al. Development of targeted therapy for ovarian cancer mediated by a plasmid expressing diphtheria toxin under the control of h19 regulatory sequences. J. Transl. Med. 2009, 7, 69. [Google Scholar] [CrossRef] [Green Version]
- Amit, D.; Hochberg, A. Development of targeted therapy for a broad spectrum of cancers (pancreatic cancer, ovarian cancer, glioblastoma and HCC) mediated by a double promoter plasmid expressing diphtheria toxin under the control of H19 and IGF2-P4 regulatory sequences. Int. J. Clin. Exp. Med. 2012, 5, 296–305. [Google Scholar]
- Sidi, A.A.; Ohana, P.; Benjamin, S.; Shalev, M.; Ransom, J.H.; Lamm, D.; Hochberg, A.; Leibovitch, I. Phase I/II marker lesion study of intravesical BC-819 DNA plasmid in H19 over expressing superficial bladder cancer refractory to bacillus calmette-guerin. J. Urol. 2008, 180, 2379–2383. [Google Scholar] [CrossRef]
- Gofrit, O.N.; Benjamin, S.; Halachmi, S.; Leibovitch, I.; Dotan, Z.; Lamm, D.L.; Ehrlich, N.; Yutkin, V.; Ben-Am, M.; Hochberg, A. DNA based therapy with diphtheria toxin-A BC-819: A phase 2b marker lesion trial in patients with intermediate risk nonmuscle invasive bladder cancer. J. Urol. 2014, 191, 1697–1702. [Google Scholar] [CrossRef]
- McCluskey, A.J.; Collier, R.J. Receptor-directed chimeric toxins created by sortase-mediated protein fusion. Mol. Cancer Ther. 2013, 12, 2273–2281. [Google Scholar] [CrossRef] [Green Version]
- Chen, H. Exploiting the intron-splicing mechanism of insect cells to produce viral vectors harboring toxic genes for suicide gene therapy. Mol. Ther. Nucleic Acids 2012, 1, e57. [Google Scholar] [CrossRef]
- Kunitomi, M.; Takayama, E.; Suzuki, S.; Yasuda, T.; Tsutsui, K.; Nagaike, K.; Hiroi, S.; Tadakuma, T. Selective inhibition of hepatoma cells using diphtheria toxin a under the control of the promoter/enhancer region of the human alpha-fetoprotein gene. Jpn. J. Cancer Res. 2000, 91, 343–350. [Google Scholar] [CrossRef]
- Gao, X.P.; Liu, Z.M.; Jiao, Y.L.; Cui, B.; Zhu, Y.T.; Zhang, J.; Wang, L.C.; Zhao, Y.R. Diphtheria toxin/human b-cell activating factor fusion protein kills human acute lymphoblastic leukemia BALL-1 cells: an experimental study. Chin. J. Cancer Res. 2012, 24, 238–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakidani, H.; Watarai, S.; Onuma, M.; Tomochika, K.; Yasuda, T. Suppressive effect of liposomes containing dna coding for diphtheria toxin A-chain on cells transformed with bovine leukemia virus. Microbiol. Immunol. 1993, 37, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Kamimura, K.; Suda, T.; Xu, W.; Zhang, G.; Liu, D. Image-guided, lobe-specific hydrodynamic gene delivery to swine liver. Mol. Ther. 2009, 17, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Kamimura, K.; Suda, T.; Zhang, G.; Aoyagi, Y.; Liu, D. Parameters affecting image-guided, hydrodynamic gene delivery to swine liver. Mol. Ther. Nucleic. Acids 2013, 2, e128. [Google Scholar] [CrossRef]
- Kamimura, K.; Kanefuji, T.; Yokoo, T.; Abe, H.; Suda, T.; Kobayashi, Y.; Zhang, G.; Aoyagi, Y.; Liu, D. Safety assessment of liver-targeted hydrodynamic gene delivery in dogs. PLoS ONE 2014, 9, e107203. [Google Scholar] [CrossRef]
- Liu, F.; Song, Y.; Liu, D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999, 6, 1258–1266. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Budker, V.; Wolff, J.A. High levels of foreign gene expression in hepatocytes after tail vein injections of naked plasmid DNA. Hum. Gene Ther. 1999, 10, 1735–1737. [Google Scholar] [CrossRef]
- Holmes, R.K. Biology and molecular epidemiology of diphtheria toxin and the tox gene. J. Infect Dis. 2000, 181 (Suppl. 1), S156–S167. [Google Scholar] [CrossRef] [Green Version]
- Bennett, M.J.; Eisenberg, D. Refined structure of monomeric diphtheria toxin at 2.3 A resolution. Protein Sci. 1994, 3, 1464–1475. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Ghani, R.; Ohana, P.; Matouk, I.; Ayesh, S.; Ayesh, B.; Laster, M.; Bibi, O.; Giladi, H.; Molnar-Kimber, K.; Sughayer, M.A.; et al. Use of transcriptional regulatory sequences of telomerase (hTER and hTERT) for selective killing of cancer cells. Mol. Ther. 2000, 2, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, K.S.; Djeha, H.A.; Gawn, J.; Cliffe, S.; Maitland, N.J.; Palmer, D.H.; Mountain, A.; Irvine, A.S.; Wrighton, C.J. Optimization of a synthetic beta-catenin-dependent promoter for tumor-specific cancer-gene therapy. Mol. Ther. 2004, 10, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Kamimura, K.; Suda, T.; Zhang, G.; Liu, D. Advances in gene delivery systems. Pharm. Med. 2011, 25, 293–306. [Google Scholar] [CrossRef]
- Kamimura, K.; Liu, D. Physical approaches for nucleic acid delivery to liver. AAPS J. 2008, 10, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Abe, H.; Kamimura, K.; Kobayashi, Y.; Ohtsuka, M.; Miura, H.; Ohashi, R.; Yokoo, T.; Kanefuji, T.; Suda, T.; Tsuchida, M.; et al. Effective prevention of liver fibrosis by liver-targeted hydrodynamic gene delivery of matrix metalloproteinase-13 in a rat liver fibrosis model. Mol. Ther. Nucleic. Acids 2016, 5, e276. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Kamimura, K.; Abe, H.; Yokoo, T.; Ogawa, K.; Shinagawa-Kobayashi, Y.; Goto, R.; Inoue, R.; Ohtsuka, M.; Miura, H.; et al. Effects of fibrotic tissue on liver-targeted hydrodynamic gene delivery. Mol. Ther. Nucleic Acids 2016, 5, e359. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Khan, S.K.; Gvozdenovic-Jeremic, J.; Kim, Y.; Dahlman, J.; Kim, H.; Park, O.; Ishitani, T.; Jho, E.H.; Gao, B.; et al. Hippo signaling interactions with wnt/beta-catenin and notch signaling repress liver tumorigenesis. J. Clin. Invest. 2017, 127, 137–152. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Lu, T.; Li, J.; Yang, R.; Hu, L.; Ye, Y.; Mao, F.; He, L.; Xu, J.; Wang, Z.; et al. The novel tumor suppressor IRF2BP2 regulates Hippo pathway in liver cancer via a feedback loop. Hepatology 2019. [Google Scholar] [CrossRef]
- Miyamura, N.; Hata, S.; Itoh, T.; Tanaka, M.; Nishio, M.; Itoh, M.; Ogawa, Y.; Terai, S.; Sakaida, I.; Suzuki, A.; et al. YAP determines the cell fate of injured mouse hepatocytes in vivo. Nat. Commun. 2017, 8, 16017. [Google Scholar] [CrossRef] [Green Version]
- Matsudaira, T.; Mukai, K.; Noguchi, T.; Hasegawa, J.; Hatta, T.; Iemura, S.I.; Natsume, T.; Miyamura, N.; Nishina, H.; Nakayama, J.; et al. Endosomal phosphatidylserine is critical for the YAP signalling pathway in proliferating cells. Nat. Commun. 2017, 8, 1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Wang, J.; Wang, H.; Fan, L.; Fan, B.; Zeng, B.; Tao, J.; Li, X.; Che, L.; Cigliano, A.; et al. Hippo cascade controls lineage commitment of liver tumors in mice and humans. Am. J. Pathol. 2018, 188, 995–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Wang, J.; Xu, Z.; Li, R.; Wang, P.; Shang, R.; Cigliano, A.; Ribback, S.; Solinas, A.; Pes, G.M.; et al. SNAI1 promotes the cholangiocellular phenotype, but not epithelial-mesenchymal transition, in a murine hepatocellular carcinoma model. Cancer Res. 2019, 79, 5563–5574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wang, H.; Peters, M.; Ding, N.; Ribback, S.; Utpatel, K.; Cigliano, A.; Dombrowski, F.; Xu, M.; Chen, X.; et al. Loss of Fbxw7 synergizes with activated Akt signaling to promote c-Myc dependent cholangiocarcinogenesis. J. Hepatol. 2019, 71, 742–752. [Google Scholar] [CrossRef]
- Nishio, M.; Sugimachi, K.; Goto, H.; Wang, J.; Morikawa, T.; Miyachi, Y.; Takano, Y.; Hikasa, H.; Itoh, T.; Suzuki, S.O.; et al. Dysregulated YAP1/TAZ and TGF-beta signaling mediate hepatocarcinogenesis in Mob1a/1b-deficient mice. Proc. Natl. Acad. Sci. USA 2016, 113, E71–E80. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Finegold, M.J.; Johnson, R.L. Hippo pathway coactivators Yap and Taz are required to coordinate mammalian liver regeneration. Exp. Mol. Med. 2018, 50, e423. [Google Scholar] [CrossRef]
- Montagner, M.; Dupont, S. Mechanical forces as determinants of disseminated metastatic cell fate. Cells 2020, 250. [Google Scholar] [CrossRef] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamimura, K.; Yokoo, T.; Abe, H.; Sakai, N.; Nagoya, T.; Kobayashi, Y.; Ohtsuka, M.; Miura, H.; Sakamaki, A.; Kamimura, H.; et al. Effect of Diphtheria Toxin-Based Gene Therapy for Hepatocellular Carcinoma. Cancers 2020, 12, 472. https://doi.org/10.3390/cancers12020472
Kamimura K, Yokoo T, Abe H, Sakai N, Nagoya T, Kobayashi Y, Ohtsuka M, Miura H, Sakamaki A, Kamimura H, et al. Effect of Diphtheria Toxin-Based Gene Therapy for Hepatocellular Carcinoma. Cancers. 2020; 12(2):472. https://doi.org/10.3390/cancers12020472
Chicago/Turabian StyleKamimura, Kenya, Takeshi Yokoo, Hiroyuki Abe, Norihiro Sakai, Takuro Nagoya, Yuji Kobayashi, Masato Ohtsuka, Hiromi Miura, Akira Sakamaki, Hiroteru Kamimura, and et al. 2020. "Effect of Diphtheria Toxin-Based Gene Therapy for Hepatocellular Carcinoma" Cancers 12, no. 2: 472. https://doi.org/10.3390/cancers12020472