
Hepatitis B Virus Epsilon (ε) RNA Element: Dynamic Regulator of Viral Replication and Attractive Therapeutic Target

 ,
,
Abstract
:1. Introduction

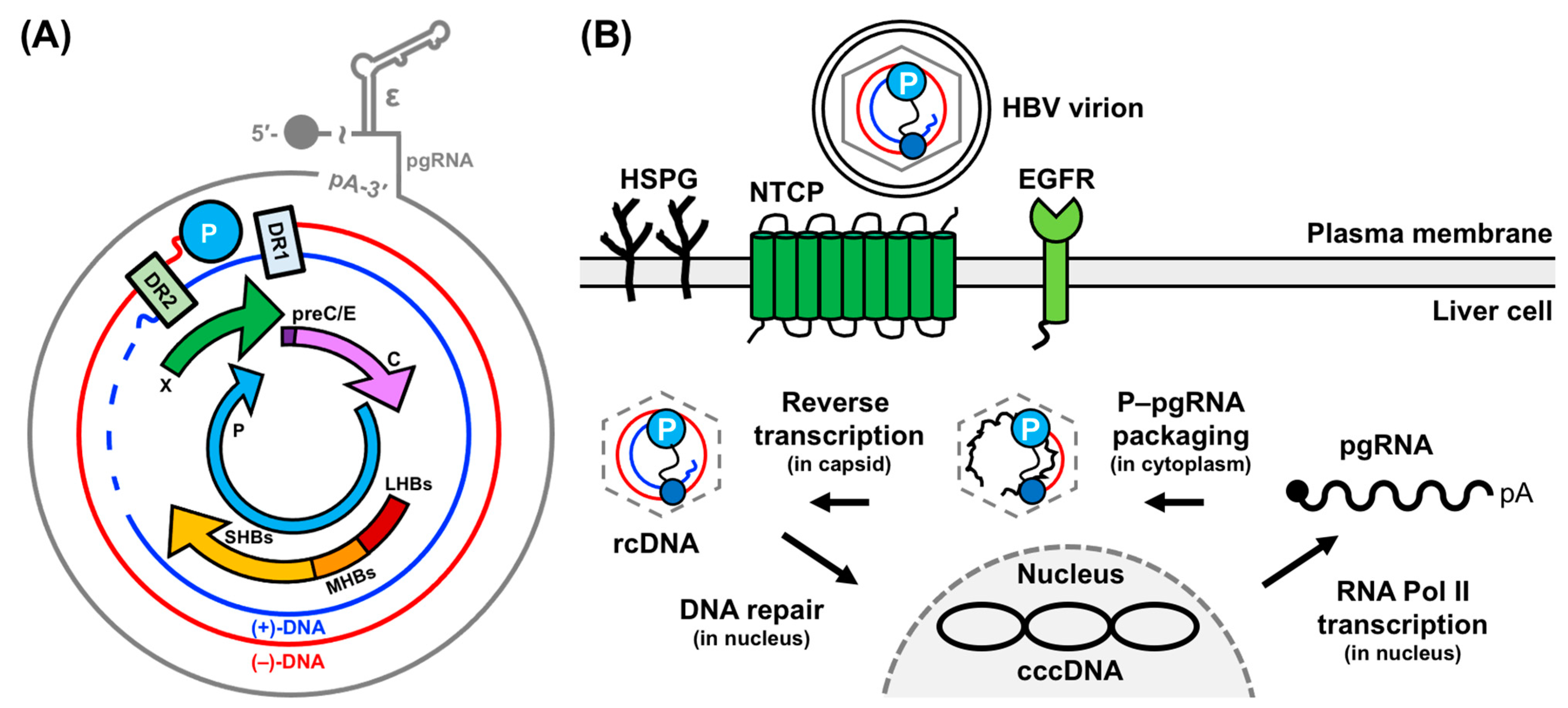
2. HBV Replication: Molecular Mechanisms and Critical Interactions
2.1. Conversion of rcDNA to cccDNA to pgRNA
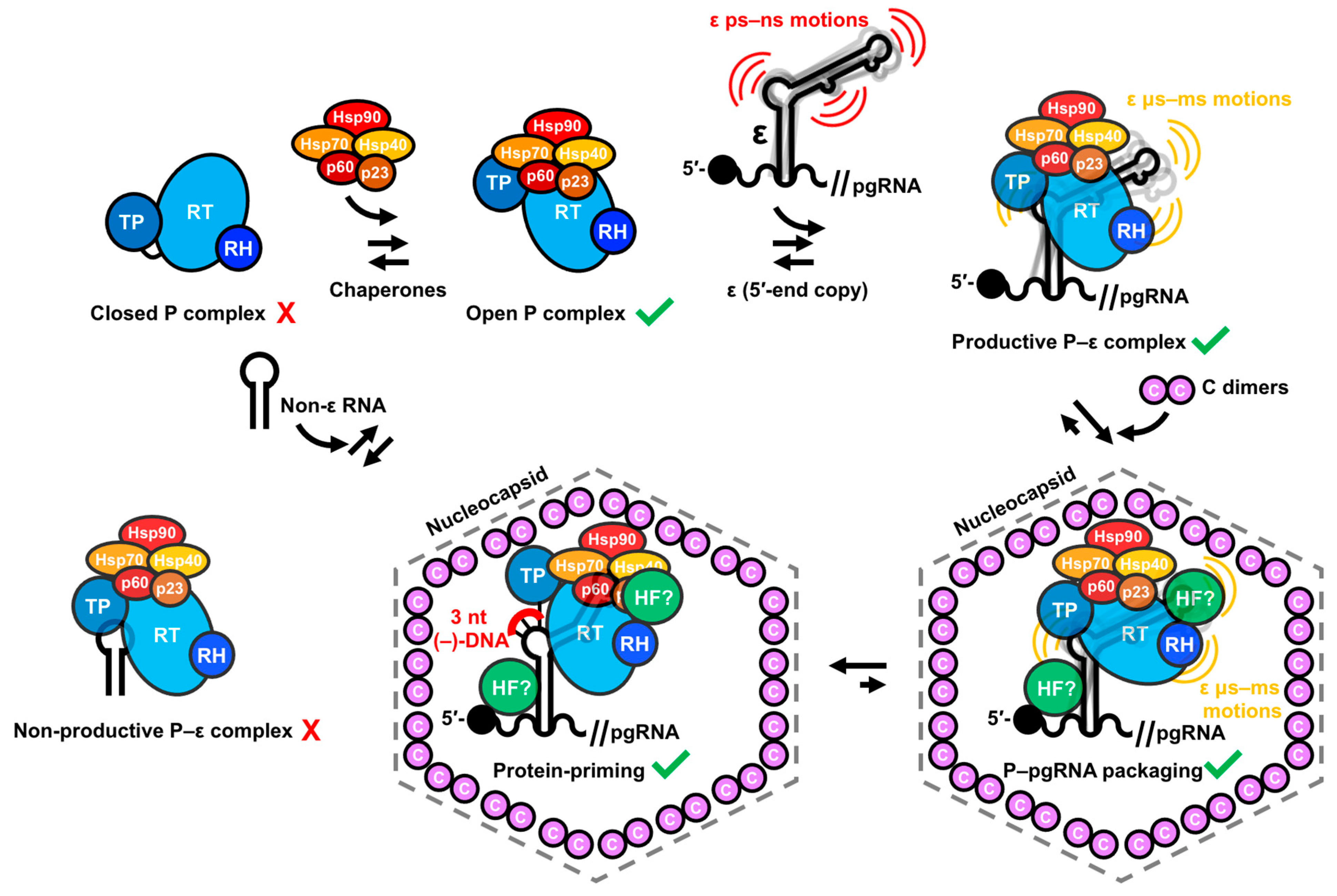
2.2. P–pgRNA Packaging and Reverse Transcription
2.3. (−)-DNA Strand Synthesis
2.4. (+)-DNA Strand Synthesis
2.5. Perspectives and Challenges to Mechanistic Studies of HBV Replication
3. Tackling HBV: Insights into Viral Replication and Evolving Therapeutic Strategies
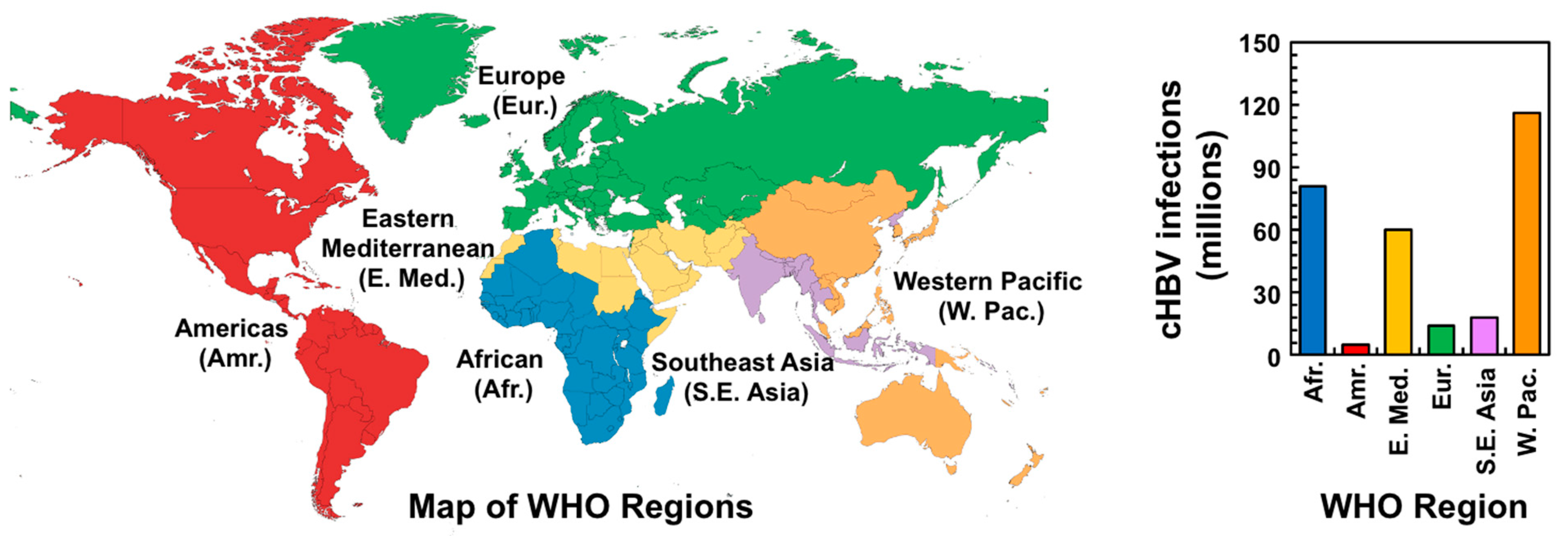
3.1. Global Burden of HBV
3.2. Current Treatments of cHBV Infection
3.2.1. FDA-Approved cHBV Treatments
NRTI Treatment

IFN-α Treatment
Perspective on Future NRTI and IFN-α Treatment
3.2.2. Alternative NRTIs in Clinical Trials for cHBV Treatment
3.3. Alternative Anti-HBV Therapies
3.3.1. Targeting Liver Cell Entry
3.3.2. Targeting the Conversion of rcDNA to cccDNA
3.3.3. Targeting the Transcriptional Activity of cccDNA
3.3.4. Targeting the ε–P Interaction
3.3.5. Targeting Protein Priming
3.3.6. Targeting RNase H Activity
3.4. HBV cccDNA: Key Obstacle for Curative HBV Treatments
4. ε as an Underexploited and Attractive Therapeutic Target
4.1. Early Characterization of ε

4.2. P Protein Structure and Host Interactions
4.3. Essential Factors and Dynamic Underpinnings of the ε–P Interaction for HBV Replication
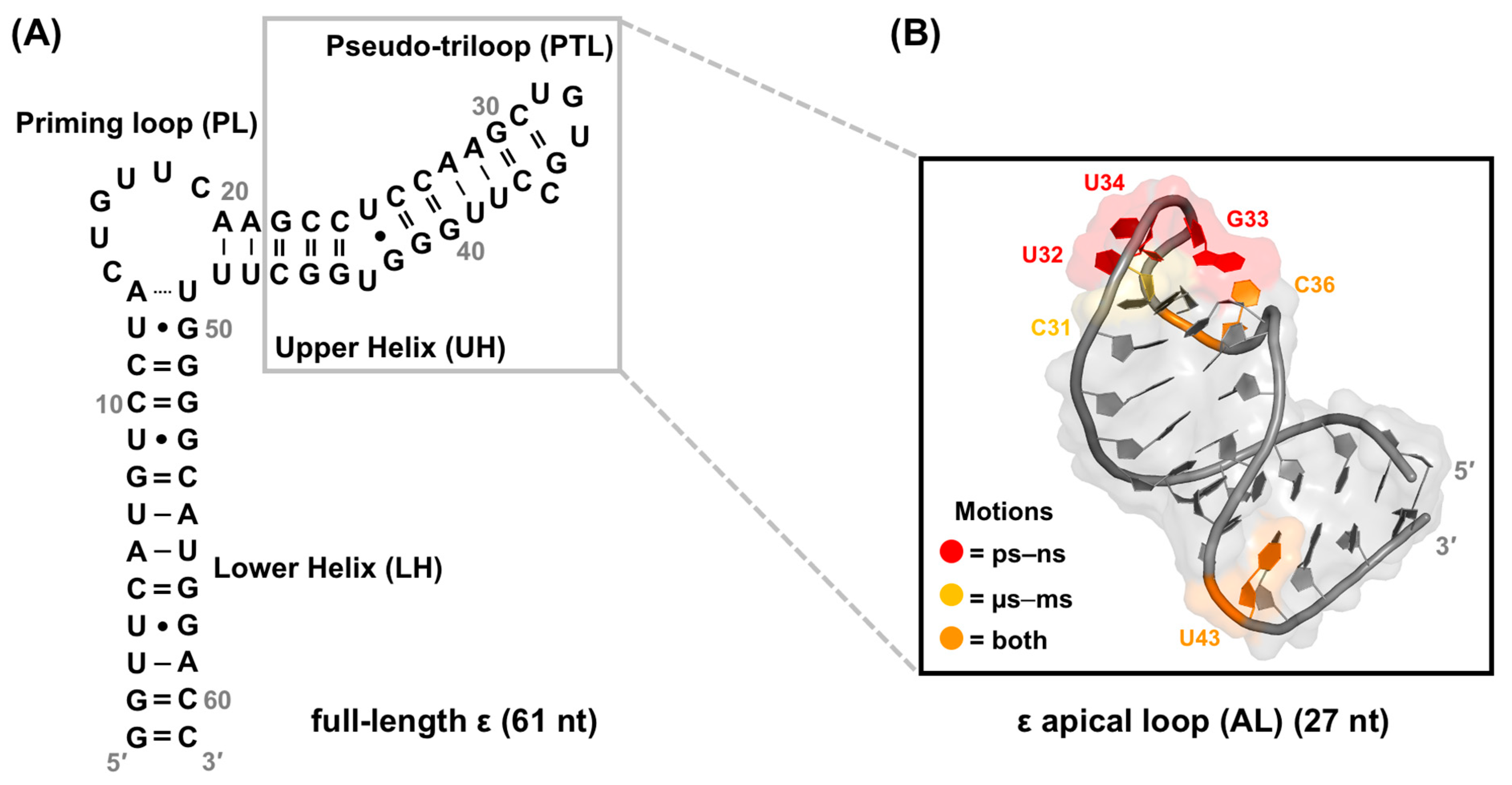
4.4. Structural Dynamics Characterization of Full-Length ε
4.5. ε Dynamics as a Regulatory Component of the ε–P Interaction in HBV Replication
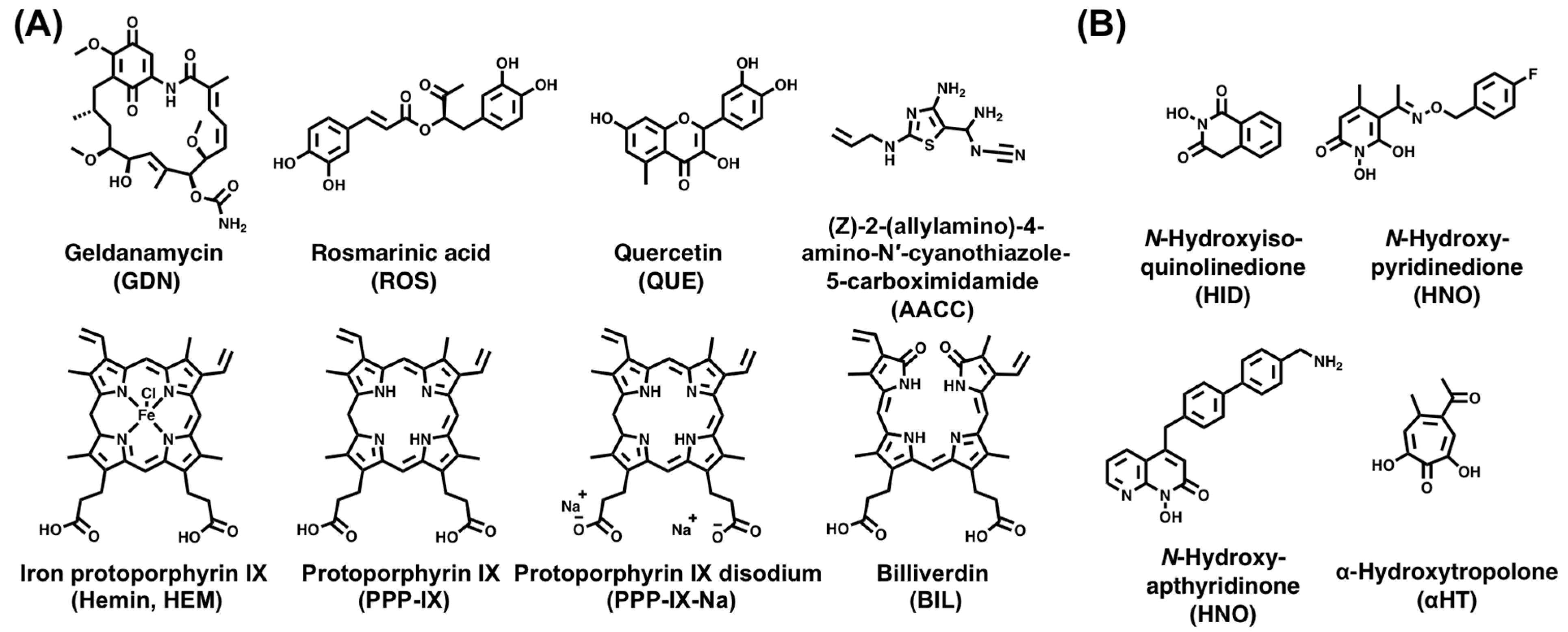
5. Discovery of ε-Targeting Small Molecules
5.1. HTS Strategy
5.2. Virtual Screen Strategy
5.3. RNA Dynamics as a Target for Future Anti-HBV Therapeutics
6. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Galibert, F.; Mandart, E.; Fitoussi, F.; Tiollais, P.; Charnay, P. Nucleotide sequence of the hepatitis B virus genome (subtype ayw) cloned in E. coli. Nature 1979, 281, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, P.; Gray, P.; Quiroga, M.; Zaldivar, J.; Goodman, H.M.; Rutter, W.J. Nucleotide sequence of the gene coding for the major protein of hepatitis B virus surface antigen. Nature 1979, 280, 815–819. [Google Scholar] [CrossRef] [PubMed]
- Charnay, P.; Mandart, E.; Hampe, A.; Fitoussi, F.; Tiollais, P.; Galibert, F. Localization on the viral genome and nucleotide sequence of the gene coding for the two major polypeptides of the Hepatitis B surface antigen (HBs Ag). Nucleic Acids Res. 1979, 7, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Pasek, M.; Goto, T.; Gilbert, W.; Zink, B.; Schaller, H.; Mackay, P.; Leadbetter, G.; Murray, K. Hepatitis B virus genes and their expression in E. coli. Nature 1979, 282, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Robinson, W.S.; Clayton, D.A.; Greenman, R.L. DNA of a Human Hepatitis B Virus Candidate. J. Virol. 1974, 14, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Hu, J. Hepatitis B virus reverse transcriptase: Diverse functions as classical and emerging targets for antiviral intervention. Emerg. Microbes Infect. 2013, 2, e56. [Google Scholar] [CrossRef]
- Bartenschlager, R.; Junker-niepmann, M.; Schaller, H. The P gene product of hepatitis B virus is required as a structural component for genomic RNA encapsidation. J. Virol. 1990, 64, 5324. [Google Scholar] [CrossRef]
- Junker-Niepmann, M.; Bartenschlager, R.; Schaller, H. A short cis-acting sequence is required for hepatitis B virus pregenome encapsidation and sufficient for packaging of foreign RNA. EMBO J. 1990, 9, 3389–3396. [Google Scholar] [CrossRef]
- Wang, G.H.; Seeger, C. The reverse transcriptase of hepatitis B virus acts as a protein primer for viral DNA synthesis. Cell 1992, 71, 663–670. [Google Scholar] [CrossRef]
- Webber, M.; Bronsema, V.; Bartos, H.; Bosserhoff, A.; Bartenschlager, R.; Schaller, H. Hepadnavirus P protein utilizes a tyrosine residue in the TP domain to prime reverse transcription. J. Virol. 1994, 68, 2994–2999. [Google Scholar] [CrossRef]
- Zoulim, F.; Seeger, C. Reverse transcription in hepatitis B viruses is primed by a tyrosine residue of the polymerase. J. Virol. 1994, 68, 6–13. [Google Scholar] [CrossRef]
- Toh, H.; Hayashida, H.; Miyata, T. Sequence homology between retroviral reverse transcriptase and putative polymerases of hepatitis B virus and cauliflower mosaic virus. Nature 1983, 305, 827–829. [Google Scholar] [CrossRef]
- Radziwill, G.; Tucker, W.; Schaller, H. Mutational analysis of the hepatitis B virus P gene product: Domain structure and RNase H activity. J. Virol. 1990, 64, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Menéndez-Arias, L.; Álvarez, M.; Pacheco, B. Nucleoside/nucleotide analog inhibitors of hepatitis B virus polymerase: Mechanism of action and resistance. Curr. Opin. Virol. 2014, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zoulim, F.; Locarnini, S. Hepatitis B virus resistance to nucleos(t)ide analogues. Gastroenterology 2009, 137, 1593–1608. [Google Scholar] [CrossRef]
- De Clercq, E.; Férir, G.; Kaptein, S.; Neyts, J. Antiviral Treatment of Chronic Hepatitis B Virus (HBV) Infections. Viruses 2010, 2, 1279–1305. [Google Scholar] [CrossRef]
- Woo, A.S.J.; Kwok, R.; Ahmed, T. Alpha-interferon treatment in hepatitis B. Ann. Transl. Med. 2017, 5, 159. [Google Scholar] [CrossRef] [PubMed]
- Scaglione, S.J.; Lok, A.S.F. Effectiveness of hepatitis B treatment in clinical practice. Gastroenterology 2012, 142, 1360–1368. [Google Scholar] [CrossRef]
- Nassal, M.; Rieger, A. A bulged region of the hepatitis B virus RNA encapsidation signal contains the replication origin for discontinuous first-strand DNA synthesis. J. Virol. 1996, 70, 2764–2773. [Google Scholar] [CrossRef]
- Wang, G.H.; Zoulim, F.; Leber, E.H.; Kitson, J.; Seeger, C. Role of RNA in enzymatic activity of the reverse transcriptase of hepatitis B viruses. J. Virol. 1994, 68, 8437–8442. [Google Scholar] [CrossRef]
- Pollack, J.R.; Ganem, D. An RNA stem-loop structure directs hepatitis B virus genomic RNA encapsidation. J. Virol. 1993, 67, 3254. [Google Scholar] [CrossRef] [PubMed]
- Knaus, T.; Nassal, M. The encapsidation signal on the hepatitis B virus RNA pregenome forms a stem-loop structure that is critical for its function. Nucleic Acids Res. 1993, 21, 3967–3975. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, R.C.; Lavine, J.E.; Chang, L.J.; Varmus, H.E.; Ganem, D. Polymerase gene products of hepatitis B viruses are required for genomic RNA packaging as wel as for reverse transcription. Nature 1990, 344, 552–555. [Google Scholar] [CrossRef]
- Rieger, A.; Nassal, M. Specific hepatitis B virus minus-strand DNA synthesis requires only the 5’ encapsidation signal and the 3’-proximal direct repeat DR1. J. Virol. 1996, 70, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Fallows, D.A.; Goff, S.P. Mutations in the epsilon sequences of human hepatitis B virus affect both RNA encapsidation and reverse transcription. J. Virol. 1995, 69, 3067–3073. [Google Scholar] [CrossRef]
- Lanford, R.E.; Notvall, L.; Beames, B. Nucleotide priming and reverse transcriptase activity of hepatitis B virus polymerase expressed in insect cells. J. Virol. 1995, 69, 4431–4439. [Google Scholar] [CrossRef]
- Bartenschlager, R.; Schaller, H. Hepadnaviral assembly is initiated by polymerase binding to the encapsidation signal in the viral RNA genome. EMBO J. 1992, 11, 3413–3420. [Google Scholar] [CrossRef]
- LeBlanc, R.M.; Kasprzak, W.K.; Longhini, A.P.; Olenginski, L.T.; Abulwerdi, F.; Ginocchio, S.; Shields, B.; Nyman, J.; Svirydava, M.; Del Vecchio, C.; et al. Structural insights of the conserved “priming loop” of hepatitis B virus pre-genomic RNA. J. Biomol. Struct. Dyn. 2022, 40, 9761–9773. [Google Scholar] [CrossRef]
- Beck, J.; Nassal, M. Hepatitis B virus replication. World J. Gastroenterol. 2007, 13, 48–64. [Google Scholar] [CrossRef]
- Zeisel, M.B.; Lucifora, J.; Mason, W.S.; Sureau, C.; Beck, J.; Levrero, M.; Kann, M.; Knolle, P.A.; Benkirane, M.; Durantel, D.; et al. Towards an HBV cure: State-of-the-art and unresolved questions--report of the ANRS workshop on HBV cure. Gut 2015, 64, 1314–1326. [Google Scholar] [CrossRef]
- Nassal, M. HBV cccDNA: Viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 2015, 64, 1972–1984. [Google Scholar] [CrossRef] [PubMed]
- Trépo, C.; Chan, H.L.Y.; Lok, A. Hepatitis B virus infection. Lancet 2014, 384, 2053–2063. [Google Scholar] [CrossRef] [PubMed]
- Yuen, M.F.; Chen, D.S.; Dusheiko, G.M.; Janssen, H.L.A.; Lau, D.T.Y.; Locarnini, S.A.; Peters, M.G.; Lai, C.L. Hepatitis B virus infection. Nat. Rev. Dis. Prim. 2018, 4, 18035. [Google Scholar] [CrossRef] [PubMed]
- Niklasch, M.; Zimmermann, P.; Nassal, M. The Hepatitis B Virus Nucleocapsid—Dynamic Compartment for Infectious Virus Production and New Antiviral Target. Biomedicines 2021, 9, 1577. [Google Scholar] [CrossRef]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012, 1, e00049. [Google Scholar] [CrossRef]
- Leistner, C.M.; Gruen-Bernhard, S.; Glebe, D. Role of glycosaminoglycans for binding and infection of hepatitis B virus. Cell Microbiol. 2008, 10, 122–133. [Google Scholar] [CrossRef]
- Schulze, A.; Gripon, P.; Urban, S. Hepatitis B virus infection initiates with a large surface protein-dependent binding to heparan sulfate proteoglycans. Hepatology 2007, 46, 1759–1768. [Google Scholar] [CrossRef]
- Iwamoto, M.; Saso, W.; Sugiyama, R.; Ishii, K.; Ohki, M.; Nagamori, S.; Suzuki, R.; Aizaki, H.; Ryo, A.; Yun, J.H.; et al. Epidermal growth factor receptor is a host-entry cofactor triggering hepatitis B virus internalization. Proc. Natl. Acad. Sci. USA 2019, 116, 8487–8492. [Google Scholar] [CrossRef]
- Iwamoto, M.; Saso, W.; Nishioka, K.; Ohashi, H.; Sugiyama, R.; Ryo, A.; Ohki, M.; Yun, J.-H.; Park, S.-Y.; Ohshima, T.; et al. The machinery for endocytosis of epidermal growth factor receptor coordinates the transport of incoming hepatitis B virus to the endosomal network. J. Biol. Chem. 2020, 295, 800–807. [Google Scholar] [CrossRef]
- Rall, L.B.; Standring, D.N.; Laub, O.; Rutter, W.J. Transcription of hepatitis B virus by RNA polymerase II. Mol. Cell. Biol. 1983, 3, 1766–1773. [Google Scholar] [CrossRef]
- Summers, J.; Mason, W.S. Replication of the genome of a hepatitis B--like virus by reverse transcription of an RNA intermediate. Cell 1982, 29, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Nassal, M. Stable HepG2-and Huh7-based human hepatoma cell lines for efficient regulated expression of infectious hepatitis B virus. J. Hepatol. 2006, 45, 636–645. [Google Scholar] [CrossRef]
- Königer, C.; Wingert, I.; Marsmann, M.; Rösler, C.; Beck, J.; Nassal, M. Involvement of the host DNA-repair enzyme TDP2 in formation of the covalently closed circular DNA persistence reservoir of hepatitis B viruses. Proc. Natl. Acad. Sci. USA 2014, 111, E4244–E4253. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Yamamoto, T.; Cullen, J.; Saputelli, J.; Aldrich, C.E.; Miller, D.S.; Litwin, S.; Furman, P.A.; Jilbert, A.R.; Mason, W.S. Kinetics of hepadnavirus loss from the liver during inhibition of viral DNA synthesis. J. Virol. 2001, 75, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Addison, W.R.; Walters, K.-A.; Wong, W.W.S.; Wilson, J.S.; Madej, D.; Jewell, L.D.; Tyrrell, D.L.J. Half-Life of the Duck Hepatitis B Virus Covalently Closed Circular DNA Pool In Vivo following Inhibition of Viral Replication. J. Virol. 2002, 76, 6356–6363. [Google Scholar] [CrossRef]
- Ganem, D.; Prince, A.M. Hepatitis B virus infection--natural history and clinical consequences. N. Engl. J. Med. 2004, 350, 1118–1129. [Google Scholar] [CrossRef]
- Gerlich, W.H.; Glebe, D.; Kramvis, A.; Magnius, L.O. Peculiarities in the designations of hepatitis B virus genes, their products, and their antigenic specificities: A potential source of misunderstandings. Virus Genes 2020, 56, 109–119. [Google Scholar] [CrossRef]
- Beck, J.; Nassal, M. Formation of a Functional Hepatitis B Virus Replication Initiation Complex Involves a Major Structural Alteration in the RNA Template. Mol. Cell. Biol. 1998, 18, 6265–6272. [Google Scholar] [CrossRef]
- Tavis, J.E.; Perri, S.; Ganem, D. Hepadnavirus reverse transcription initiates within the stem-loop of the RNA packaging signal and employs a novel strand transfer. J. Virol. 1994, 68, 3536–3543. [Google Scholar] [CrossRef]
- Jeong, J.-K.; Yoon, G.-S.; Ryu, W.-S. Evidence that the 5’-end cap structure is essential for encapsidation of hepatitis B virus pregenomic RNA. J. Virol. 2000, 74, 5502–5508. [Google Scholar] [CrossRef]
- Mangus, D.A.; Evans, M.C.; Jacobson, A. Poly(A)-binding proteins: Multifunctional scaffolds for the post-transcriptional control of gene expression. Genome Biol. 2003, 4, 223. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; McLachlan, A. A pregenomic RNA sequence adjacent to DR1 and complementary to epsilon influences hepatitis B virus replication efficiency. Virology 2002, 303, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.-K.; Lee, J.; Ryu, W.-S. A Novel cis-Acting Element Facilitates Minus-Strand DNA Synthesis during Reverse Transcription of the Hepatitis B Virus Genome. J. Virol. 2004, 78, 6252. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Ji, L.; Maguire, M.L.; Loeb, D.D. cis-Acting sequences that contribute to the synthesis of relaxed-circular DNA of human hepatitis B virus. J. Virol. 2004, 78, 642–649. [Google Scholar] [CrossRef]
- Abraham, T.M.; Loeb, D.D. Base pairing between the 5’ half of epsilon and a cis-acting sequence, phi, makes a contribution to the synthesis of minus-strand DNA for human hepatitis B virus. J. Virol. 2006, 80, 4380–4387. [Google Scholar] [CrossRef]
- Kamtekar, S.; Berman, A.J.; Wang, J.; Lázaro, J.M.; De Vega, M.; Blanco, L.; Salas, M.; Steitz, T.A. The φ29 DNA polymerase:protein-primer structure suggests a model for the initiation to elongation transition. EMBO J. 2006, 25, 1335–1343. [Google Scholar] [CrossRef]
- Loeb, D.D.; Hirsch, R.C.; Ganem, D. Sequence-independent RNA cleavages generate the primers for plus strand DNA synthesis in hepatitis B viruses: Implications for other reverse transcribing elements. EMBO J. 1991, 10, 3533–3540. [Google Scholar] [CrossRef]
- Lee, J.; Shin, M.K.; Lee, H.J.; Yoon, G.; Ryu, W.S. Three novel cis-acting elements required for efficient plus-strand DNA synthesis of the hepatitis B virus genome. J. Virol. 2004, 78, 7455–7464. [Google Scholar] [CrossRef]
- Wynne, S.A.; Crowther, R.A.; Leslie, A.G.W. The crystal structure of the human hepatitis B virus capsid. Mol. Cell 1999, 3, 771–780. [Google Scholar] [CrossRef]
- Kappel, K.; Zhang, K.; Su, Z.; Watkins, A.M.; Kladwang, W.; Li, S.; Pintilie, G.; Topkar, V.V.; Rangan, R.; Zheludev, I.N.; et al. Accelerated cryo-EM-guided determination of three-dimensional RNA-only structures. Nat. Methods 2020, 17, 699–707. [Google Scholar] [CrossRef]
- Chauvier, A.; Porta, J.C.; Deb, I.; Ellinger, E.; Meze, K.; Frank, A.T.; Ohi, M.D.; Walter, N.G. Structural basis for control of bacterial RNA polymerase pausing by a riboswitch and its ligand. Nat. Struct. Mol. Biol. 2023, 30, 902–913. [Google Scholar] [CrossRef]
- Guaita, M.; Watters, S.C.; Loerch, S. Recent advances and current trends in cryo-electron microscopy. Curr. Opin. Struct. Biol. 2022, 77, 102484. [Google Scholar] [CrossRef] [PubMed]
- Sheena, B.S.; Hiebert, L.; Han, H.; Ippolito, H.; Abbasi-Kangevari, M.; Abbasi-Kangevari, Z.; Abbastabar, H.; Abdoli, A.; Abubaker Ali, H.; Adane, M.M.; et al. Global, regional, and national burden of hepatitis B, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet. Gastroenterol. Hepatol. 2022, 7, 796–829. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Hepatitis B vaccines. Wkly. Epidemiol. Rec. 2004, 79, 253–264. [Google Scholar]
- Stanaway, J.D.; Flaxman, A.D.; Naghavi, M.; Fitzmaurice, C.; Vos, T.; Abubakar, I.; Abu-Raddad, L.J.; Assadi, R.; Bhala, N.; Cowie, B.; et al. The global burden of viral hepatitis from 1990 to 2013: Findings from the Global Burden of Disease Study 2013. Lancet 2016, 388, 1081–1088. [Google Scholar] [CrossRef]
- Perz, J.F.; Armstrong, G.L.; Farrington, L.A.; Hutin, Y.J.F.; Bell, B.P. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J. Hepatol. 2006, 45, 529–538. [Google Scholar] [CrossRef]
- Ioannou, G.N. Hepatitis B virus in the United States: Infection, exposure, and immunity rates in a nationally representative survey. Ann. Intern. Med. 2011, 154, 319–328. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Screening for Chronic Hepatitis B Among Asian/Pacific Islander Populations-New York City, 2005. Morb. Mortal. Wkly. Rep. 2006, 55, 505–509. [Google Scholar]
- Hatzakis, A.; Wait, S.; Bruix, J.; Buti, M.; Carballo, M.; Cavaleri, M.; Colombo, M.; Delarocque-Astagneau, E.; Dusheiko, G.; Esmat, G.; et al. The state of hepatitis B and C in Europe: Report from the hepatitis B and C summit conference*. J. Viral Hepat. 2011, 18, 1–16. [Google Scholar] [CrossRef]
- Gill, U.S.; Peppa, D.; Micco, L.; Singh, H.D.; Carey, I.; Foster, G.R.; Maini, M.K.; Kennedy, P.T.F. Interferon Alpha Induces Sustained Changes in NK Cell Responsiveness to Hepatitis B Viral Load Suppression In Vivo. PLoS Pathog. 2016, 12, e1005788. [Google Scholar] [CrossRef]
- Micco, L.; Peppa, D.; Loggi, E.; Schurich, A.; Jefferson, L.; Cursaro, C.; Panno, A.M.; Bernardi, M.; Brander, C.; Bihl, F.; et al. Differential boosting of innate and adaptive antiviral responses during pegylated-interferon-alpha therapy of chronic hepatitis B. J. Hepatol. 2013, 58, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Marcellin, P.; Heathcote, E.J.; Buti, M.; Gane, E.; de Man, R.A.; Krastev, Z.; Germanidis, G.; Lee, S.S.; Flisiak, R.; Kaita, K.; et al. Tenofovir Disoproxil Fumarate versus Adefovir Dipivoxil for Chronic Hepatitis B. N. Engl. J. Med. 2008, 359, 2442–2455. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.-L.; Gane, E.; Liaw, Y.-F.; Hsu, C.-W.; Thongsawat, S.; Wang, Y.; Chen, Y.; Heathcote, E.J.; Rasenack, J.; Bzowej, N.; et al. Telbivudine versus Lamivudine in Patients with Chronic Hepatitis B. N. Engl. J. Med. 2009, 357, 2576–2588. [Google Scholar] [CrossRef]
- Dienstag, J.L.; Schiff, E.R.; Wright, T.L.; Perrillo, R.P.; Hann, H.-W.L.; Goodman, Z.; Crowther, L.; Condreay, L.D.; Woessner, M.; Rubin, M.; et al. Lamivudine as initial treatment for chronic hepatitis B in the United States. N. Engl. J. Med. 1999, 341, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Marcellin, P.; Chang, T.-T.; Lim, S.G.; Tong, M.J.; Sievert, W.; Shiffman, M.L.; Jeffers, L.; Goodman, Z.; Wulfsohn, M.S.; Xiong, S.; et al. Adefovir dipivoxil for the treatment of hepatitis B e antigen-positive chronic hepatitis B. N. Engl. J. Med. 2003, 348, 808–816. [Google Scholar] [CrossRef]
- Chang, T.-T.; Gish, R.G.; de Man, R.; Gadano, A.; Sollano, J.; Chao, Y.-C.; Lok, A.S.; Han, K.-H.; Goodman, Z.; Zhu, J.; et al. A comparison of entecavir and lamivudine for HBeAg-positive chronic hepatitis B. N. Engl. J. Med. 2006, 354, 1001–1010. [Google Scholar] [CrossRef]
- De Zeng, M.; Mao, Y.M.; Yao, G.B.; Wang, H.; Hou, J.L.; Wang, Y.Z.; Ji, B.N.; Chang, C.N.P.; Barker, K.F. A double-blind randomized trial of adefovir dipivoxil in Chinese subjects with HBeAg-positive chronic hepatitis B. Hepatology 2006, 44, 108–116. [Google Scholar] [CrossRef]
- Lai, C.-L.; Shouval, D.; Lok, A.S.; Chang, T.-T.; Cheinquer, H.; Goodman, Z.; DeHertogh, D.; Wilber, R.; Zink, R.C.; Cross, A.; et al. Entecavir versus Lamivudine for Patients with HBeAg-Negative Chronic Hepatitis B. N. Engl. J. Med. 2009, 354, 1011–1020. [Google Scholar] [CrossRef]
- Hadziyannis, S.J.; Tassopoulos, N.C.; Heathcote, E.J.; Chang, T.-T.; Kitis, G.; Rizzetto, M.; Marcellin, P.; Lim, S.G.; Goodman, Z.; Wulfsohn, M.S.; et al. Adefovir Dipivoxil for the Treatment of Hepatitis B e Antigen–Negative Chronic Hepatitis B. N. Engl. J. Med. 2009, 348, 800–807. [Google Scholar] [CrossRef]
- Lok, A.S.F.; Lai, C.L.; Leung, N.; Yao, G.B.; Cui, Z.Y.; Schiff, E.R.; Dienstag, J.L.; Heathcote, E.J.; Little, N.R.; Griffiths, D.A.; et al. Long-term safety of lamivudine treatment in patients with chronic hepatitis B. Gastroenterology 2003, 125, 1714–1722. [Google Scholar] [CrossRef]
- Lai, C.-L.; Chien, R.-N.; Leung, N.W.Y.; Chang, T.-T.; Guan, R.; Tai, D.-I.; Ng, K.-Y.; Wu, P.-C.; Dent, J.C.; Barber, J.; et al. A one-year trial of lamivudine for chronic hepatitis B. Asia Hepatitis Lamivudine Study Group. N. Engl. J. Med. 1998, 339, 61–68. [Google Scholar] [CrossRef]
- Kayaaslan, B.; Guner, R. Adverse effects of oral antiviral therapy in chronic hepatitis B. World J. Hepatol. 2017, 9, 227–241. [Google Scholar] [CrossRef]
- Radhakrishnan, R.; Walter, L.J.; Hruza, A.; Reichert, P.; Trotta, P.P.; Nagabhushan, T.L.; Walter, M.R. Zinc mediated dimer of human interferon-alpha 2b revealed by X-ray crystallography. Structure 1996, 4, 1453–1463. [Google Scholar] [CrossRef]
- Reizis, B.; Bunin, A.; Ghosh, H.S.; Lewis, K.L.; Sisirak, V. Plasmacytoid dendritic cells: Recent progress and open questions. Annu. Rev. Immunol. 2011, 29, 163–183. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, A.; Lindenmann, J.; Valentine, R.C. Virus interference. II. Some properties of interferon. Proc. R. Soc. Lond. Ser. B Biol. Sci. 1957, 147, 268–273. [Google Scholar] [CrossRef]
- Isaacs, A.; Lindenmann, J. Virus interference. I. The interferon. Proc. R. Soc. Lond. Ser. B Biol. Sci. 1957, 147, 258–267. [Google Scholar] [CrossRef]
- Lucifora, J.; Xia, Y.; Reisinger, F.; Zhang, K.; Stadler, D.; Cheng, X.; Sprinzl, M.F.; Koppensteiner, H.; Makowska, Z.; Volz, T.; et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 2014, 343, 1221–1228. [Google Scholar] [CrossRef]
- Xu, F.; Song, H.; Li, N.; Tan, G. HBsAg blocks TYPE I IFN induced up-regulation of A3G through inhibition of STAT3. Biochem. Biophys. Res. Commun. 2016, 473, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Turelli, P.; Mangeat, B.; Jost, S.; Vianin, S.; Trono, D. Inhibition of hepatitis B virus replication by APOBEC3G. Science 2004, 303, 1829. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.H.; Gummuluru, S.; Hu, J. Deamination-Independent Inhibition of Hepatitis B Virus Reverse Transcription by APOBEC3G. J. Virol. 2007, 81, 4465–4472. [Google Scholar] [CrossRef]
- Belloni, L.; Allweiss, L.; Guerrieri, F.; Pediconi, N.; Volz, T.; Pollicino, T.; Petersen, J.; Raimondo, G.; Dandri, M.; Levrero, M. IFN-α inhibits HBV transcription and replication in cell culture and in humanized mice by targeting the epigenetic regulation of the nuclear cccDNA minichromosome. J. Clin. Invest. 2012, 122, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.L.Y.; Leung, N.W.Y.; Hui, A.Y.; Wong, V.W.S.; Liew, C.T.; Chim, A.M.L.; Chan, F.K.L.; Hung, L.C.T.; Lee, Y.T.; Tam, J.S.L.; et al. A randomized, controlled trial of combination therapy for chronic hepatitis B: Comparing pegylated interferon-alpha2b and lamivudine with lamivudine alone. Ann. Intern. Med. 2005, 142, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Janssen, H.L.A.; Van Zonneveld, M.; Senturk, H.; Zeuzem, S.; Akarca, U.S.; Cakaloglu, Y.; Simon, C.; So, T.M.K.; Gerken, G.; de Man, R.A.; et al. Pegylated interferon alfa-2b alone or in combination with lamivudine for HBeAg-positive chronic hepatitis B: A randomised trial. Lancet 2005, 365, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Lau, G.K.K.; Piratvisuth, T.; Luo, K.X.; Marcellin, P.; Thongsawat, S.; Cooksley, G.; Gane, E.; Fried, M.W.; Chow, W.C.; Paik, S.W.; et al. Peginterferon Alfa-2a, lamivudine, and the combination for HBeAg-positive chronic hepatitis B. N. Engl. J. Med. 2005, 352, 2682–2695. [Google Scholar] [CrossRef]
- Marcellin, P.; Lau, G.K.K.; Bonino, F.; Farci, P.; Hadziyannis, S.; Jin, R.; Lu, Z.-M.; Piratvisuth, T.; Germanidis, G.; Yurdaydin, C.; et al. Peginterferon alfa-2a alone, lamivudine alone, and the two in combination in patients with HBeAg-negative chronic hepatitis B. N. Engl. J. Med. 2004, 351, 1206–1217. [Google Scholar] [CrossRef]
- Wong, V.W.S.; Wong, G.L.H.; Yan, K.K.L.; Chim, A.M.L.; Chan, H.Y.; Tse, C.H.; Choi, P.C.L.; Chan, A.W.H.; Sung, J.J.Y.; Chan, H.L.Y. Durability of peginterferon alfa-2b treatment at 5 years in patients with hepatitis B e antigen–positive chronic hepatitis B. Hepatology 2010, 51, 1945–1953. [Google Scholar] [CrossRef]
- Buster, E.H.C.J.; Flink, H.J.; Cakaloglu, Y.; Simon, K.; Trojan, J.; Tabak, F.; So, T.M.K.; Feinman, S.V.; Mach, T.; Akarca, U.S.; et al. Sustained HBeAg and HBsAg loss after long-term follow-up of HBeAg-positive patients treated with peginterferon alpha-2b. Gastroenterology 2008, 135, 459–467. [Google Scholar] [CrossRef]
- Koumbi, L. Current and future antiviral drug therapies of hepatitis B chronic infection. World J. Hepatol. 2015, 7, 1030–1040. [Google Scholar] [CrossRef] [PubMed]
- Monto, A.; Schooley, R.T.; Lai, J.C.; Sulkowski, M.S.; Chung, R.T.; Pawlotsky, J.M.; McHutchison, J.G.; Jacobson, I.M. Lessons from HIV therapy applied to viral hepatitis therapy: Summary of a workshop. Am. J. Gastroenterol. 2010, 105, 989–1004. [Google Scholar] [CrossRef]
- Ghany, M.; Liang, T.J. Drug Targets and Molecular Mechanisms of Drug Resistance in Chronic Hepatitis B. Gastroenterology 2007, 132, 1574–1585. [Google Scholar] [CrossRef]
- Takkenberg, B.; Terpstra, V.; Zaaijer, H.; Weegink, C.; Dijkgraaf, M.; Jansen, P.; Beld, M.; Reesink, H. Intrahepatic response markers in chronic hepatitis B patients treated with peginterferon alpha-2a and adefovir. J. Gastroenterol. Hepatol. 2011, 26, 1527–1535. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.L.; Ahn, S.H.; Lee, K.S.; Um, S.H.; Cho, M.; Yoon, S.K.; Lee, J.W.; Park, N.H.; Kweon, Y.O.; Sohn, J.H.; et al. Phase IIb multicentred randomised trial of besifovir (LB80380) versus entecavir in Asian patients with chronic hepatitis B. Gut 2014, 63, 996–1004. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.H.; Kim, W.; Jung, Y.K.; Yang, J.M.; Jang, J.Y.; Kweon, Y.O.; Cho, Y.K.; Kim, Y.J.; Hong, G.Y.; Kim, D.J.; et al. Efficacy and Safety of Besifovir Dipivoxil Maleate Compared With Tenofovir Disoproxil Fumarate in Treatment of Chronic Hepatitis B Virus Infection. Clin. Gastroenterol. Hepatol. 2019, 17, 1850–1859.e4. [Google Scholar] [CrossRef] [PubMed]
- Yuen, M.F.; Ahn, S.H.; Lee, K.S.; Um, S.H.; Cho, M.; Yoon, S.K.; Lee, J.W.; Park, N.H.; Kweon, Y.O.; Sohn, J.H.; et al. Two-year treatment outcome of chronic hepatitis B infection treated with besifovir vs. entecavir: Results from a multicentre study. J. Hepatol. 2015, 62, 526–532. [Google Scholar] [CrossRef]
- Painter, G.R.; Almond, M.R.; Trost, L.C.; Lampert, B.M.; Neyts, J.; De Clercq, E.; Korba, B.E.; Aldern, K.A.; Beadle, J.R.; Hostetler, K.Y. Evaluation of hexadecyloxypropyl-9-R-[2-(phosphonomethoxy)propyl]-adenine, CMX157, as a potential treatment for human immunodeficiency virus type 1 and hepatitis B virus infections. Antimicrob. Agents Chemother. 2007, 51, 3505–3509. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Park, K.S.; Kim, N.H.; Cho, J.Y.; Koh, M.S.; Lee, J.H. Clevudine Induced Mitochondrial Myopathy. J. Korean Med. Sci. 2017, 32, 1857–1860. [Google Scholar] [CrossRef]
- Jones, S.A.; Murakami, E.; Delaney, W.; Furman, P.; Hu, J. Noncompetitive Inhibition of Hepatitis B Virus Reverse Transcriptase Protein Priming and DNA Synthesis by the Nucleoside Analog Clevudine. Antimicrob. Agents Chemother. 2013, 57, 4181–4189. [Google Scholar] [CrossRef]
- Niu, C.; Murakami, E.; Furman, P.A. Clevudine is Efficiently Phosphorylated to the Active Triphosphate form in Primary Human Hepatocytes. Antivir. Ther. 2008, 13, 263–269. [Google Scholar] [CrossRef]
- Anderson, D.L. Clevudine for hepatitis B. Drugs Today 2009, 45, 331–350. [Google Scholar] [CrossRef]
- Squires, K.E.; Mayers, D.L.; Bluemling, G.R.; Kolykhalov, A.A.; Guthrie, D.B.; Reddy, P.; Mitchell, D.G.; Saindane, M.T.; Sticher, Z.M.; Edpuganti, V.; et al. ATI-2173, a Novel Liver-Targeted Non-Chain-Terminating Nucleotide for Hepatitis B Virus Cure Regimens. Antimicrob. Agents Chemother. 2020, 64, 10–1128. [Google Scholar] [CrossRef]
- Squires, K.E.; Ogilvie, L.; Jucov, A.; Anastasiy, I.; Ghicavii, N.; Huguet, J.; Melara, R.; Constantineau, M.; De La Rosa, A.; Mayers, D.L. A randomized phase 1b trial of the active site polymerase inhibitor nucleotide ATI-2173 in patients with chronic hepatitis B virus infection. J. Viral Hepat. 2023, 30, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Lempp, F.A.; Urban, S. Inhibitors of hepatitis B virus attachment and entry. Intervirology 2014, 57, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Verrier, E.R.; Schuster, C.; Baumert, T.F. Advancing hepatitis B virus entry inhibitors. J. Hepatol. 2017, 66, 677–679. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, J.; Li, T. Regulation of the HBV Entry Receptor NTCP and its Potential in Hepatitis B Treatment. Front. Mol. Biosci. 2022, 9, 879817. [Google Scholar] [CrossRef] [PubMed]
- Gripon, P.; Cannie, I.; Urban, S. Efficient inhibition of hepatitis B virus infection by acylated peptides derived from the large viral surface protein. J. Virol. 2005, 79, 1613–1622. [Google Scholar] [CrossRef]
- Schulze, A.; Schieck, A.; Ni, Y.; Mier, W.; Urban, S. Fine mapping of pre-S sequence requirements for hepatitis B virus large envelope protein-mediated receptor interaction. J. Virol. 2010, 84, 1989–2000. [Google Scholar] [CrossRef] [PubMed]
- Petersen, J.; Dandri, M.; Mier, W.; Lütgehetmann, M.; Volz, T.; Von Weizsäcker, F.; Haberkorn, U.; Fischer, L.; Pollok, J.M.; Erbes, B.; et al. Prevention of hepatitis B virus infection in vivo by entry inhibitors derived from the large envelope protein. Nat. Biotechnol. 2008, 26, 335–341. [Google Scholar] [CrossRef]
- Volz, T.; Allweiss, L.; ḾBarek, M.B.; Warlich, M.; Lohse, A.W.; Pollok, J.M.; Alexandrov, A.; Urban, S.; Petersen, J.; Lütgehetmann, M.; et al. The entry inhibitor Myrcludex-B efficiently blocks intrahepatic virus spreading in humanized mice previously infected with hepatitis B virus. J. Hepatol. 2013, 58, 861–867. [Google Scholar] [CrossRef]
- Vaz, F.M.; Paulusma, C.C.; Huidekoper, H.; de Ru, M.; Lim, C.; Koster, J.; Ho-Mok, K.; Bootsma, A.H.; Groen, A.K.; Schaap, F.G.; et al. Sodium taurocholate cotransporting polypeptide (SLC10A1) deficiency: Conjugated hypercholanemia without a clear clinical phenotype. Hepatology 2015, 61, 260–267. [Google Scholar] [CrossRef]
- Bogomolov, P.; Alexandrov, A.; Voronkova, N.; Macievich, M.; Kokina, K.; Petrachenkova, M.; Lehr, T.; Lempp, F.A.; Wedemeyer, H.; Haag, M.; et al. Treatment of chronic hepatitis D with the entry inhibitor myrcludex B: First results of a phase Ib/IIa study. J. Hepatol. 2016, 65, 490–498. [Google Scholar] [CrossRef]
- Asami, J.; Kimura, K.T.; Fujita-Fujiharu, Y.; Ishida, H.; Zhang, Z.; Nomura, Y.; Liu, K.; Uemura, T.; Sato, Y.; Ono, M.; et al. Structure of the bile acid transporter and HBV receptor NTCP. Nature 2022, 606, 1021–1026. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Iwamoto, M.; Yun, J.H.; Uchikubo-Kamo, T.; Son, D.; Jin, Z.; Yoshida, H.; Ohki, M.; Ishimoto, N.; Mizutani, K.; et al. Structural insights into the HBV receptor and bile acid transporter NTCP. Nature 2022, 606, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Mills, C.; Yu, W.; Yan, R.; Aldrich, C.E.; Saputelli, J.R.; Mason, W.S.; Xu, X.; Guo, J.T.; Block, T.M.; et al. Identification of disubstituted sulfonamide compounds as specific inhibitors of hepatitis B virus covalently closed circular DNA formation. Antimicrob. Agents Chemother. 2012, 56, 4277–4288. [Google Scholar] [CrossRef]
- Yuen, M.F.; Gane, E.J.; Kim, D.J.; Weilert, F.; Yuen Chan, H.L.; Lalezari, J.; Hwang, S.G.; Nguyen, T.; Flores, O.; Hartman, G.; et al. Antiviral Activity, Safety, and Pharmacokinetics of Capsid Assembly Modulator NVR 3-778 in Patients with Chronic HBV Infection. Gastroenterology 2019, 156, 1392–1403.e7. [Google Scholar] [CrossRef]
- Lam, A.M.; Espiritu, C.; Vogel, R.; Ren, S.; Lau, V.; Kelly, M.; Kuduk, S.D.; Hartman, G.D.; Flores, O.A.; Klumpp, K. Preclinical Characterization of NVR 3-778, a First-in-Class Capsid Assembly Modulator against Hepatitis B Virus. Antimicrob. Agents Chemother. 2018, 63, e01734-18. [Google Scholar] [CrossRef] [PubMed]
- Berke, J.M.; Dehertogh, P.; Vergauwen, K.; Mostmans, W.; Vandyck, K.; Raboisson, P.; Pauwels, F. Antiviral Properties and Mechanism of Action Studies of the Hepatitis B Virus Capsid Assembly Modulator JNJ-56136379. Antimicrob. Agents Chemother. 2020, 64, e02439-19. [Google Scholar] [CrossRef]
- Zoulim, F.; Lenz, O.; Vandenbossche, J.J.; Talloen, W.; Verbinnen, T.; Moscalu, I.; Streinu-Cercel, A.; Bourgeois, S.; Buti, M.; Crespo, J.; et al. JNJ-56136379, an HBV Capsid Assembly Modulator, Is Well-Tolerated and Has Antiviral Activity in a Phase 1 Study of Patients With Chronic Infection. Gastroenterology 2020, 159, 521–533. [Google Scholar] [CrossRef]
- Huang, Q.; Cai, D.; Yan, R.; Li, L.; Zong, Y.; Guo, L.; Mercier, A.; Zhou, Y.; Tang, A.; Henne, K.; et al. Preclinical Profile and Characterization of the Hepatitis B Virus Core Protein Inhibitor ABI-H0731. Antimicrob. Agents Chemother. 2020, 64, e01463-20. [Google Scholar] [CrossRef]
- Yuen, M.F.; Agarwal, K.; Gane, E.J.; Schwabe, C.; Ahn, S.H.; Kim, D.J.; Lim, Y.S.; Cheng, W.; Sievert, W.; Visvanathan, K.; et al. Safety, pharmacokinetics, and antiviral effects of ABI-H0731, a hepatitis B virus core inhibitor: A randomised, placebo-controlled phase 1 trial. Lancet Gastroenterol. Hepatol. 2020, 5, 152–166. [Google Scholar] [CrossRef]
- Yuen, M.F.; Zhou, X.; Gane, E.; Schwabe, C.; Tanwandee, T.; Feng, S.; Jin, Y.; Triyatni, M.; Lemenuel-Diot, A.; Cosson, V.; et al. Safety, pharmacokinetics, and antiviral activity of RO7049389, a core protein allosteric modulator, in patients with chronic hepatitis B virus infection: A multicentre, randomised, placebo-controlled, phase 1 trial. Lancet Gastroenterol. Hepatol. 2021, 6, 723–732. [Google Scholar] [CrossRef]
- Feng, S.; Gane, E.; Schwabe, C.; Zhu, M.; Triyatni, M.; Zhou, J.; Bo, Q.; Jin, Y. A Five-in-One First-in-Human Study To Assess Safety, Tolerability, and Pharmacokinetics of RO7049389, an Inhibitor of Hepatitis B Virus Capsid Assembly, after Single and Multiple Ascending Doses in Healthy Participants. Antimicrob. Agents Chemother. 2020, 64, e01323-20. [Google Scholar] [CrossRef] [PubMed]
- Mani, N.; Cole, A.G.; Phelps, J.R.; Ardzinski, A.; Burns, R.; Chiu, T.; Cuconati, A.; Dorsey, B.D.; Evangelista, E.; Fan, K.; et al. Preclinical characterization of AB-506, an inhibitor of HBV replication targeting the viral core protein. Antivir. Res. 2022, 197, 105211. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Mai, J.; Wu, M.; Chen, H.; Li, X.; Li, C.; Liu, J.; Liu, C.; Hu, Y.; Zhu, X.; et al. Safety, tolerability, pharmacokinetics, and antiviral activity of the novel core protein allosteric modulator ZM-H1505R (Canocapavir) in chronic hepatitis B patients: A randomized multiple-dose escalation trial. BMC Med. 2023, 21, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yeh, M.-L.; Chen, C.; Cheng, P.-N.; Pai, M.-C.; Chen, J.-J.; Lo, C.; Tai, C.-M.; Tsai, C.-Y.; Tseng, K.-C.; Chen, C.-H.; et al. Efficacy and safety of GLS4/ritonavir combined with entecavir in HBeAg-positive patients with chronic hepatitis B: Interim results from phase 2b, multi-center study. J. Hepatol. 2020, 73, S878–S880. [Google Scholar] [CrossRef]
- Janssen, H.; Hou, J.; Asselah, T.; Chan, H.; Zoulim, F.; Tanaka, Y.; Janczewska, E.; Nahass, R.; Bourgeois, S.; Buti, M.; et al. Efficacy and safety results of the phase 2 JNJ-56136379 JADE study in patients with chronic hepatitis B: Interim week 24 data. J. Hepatol. 2020, 73, S129–S130. [Google Scholar] [CrossRef]
- Liu, F.; Campagna, M.; Qi, Y.; Zhao, X.; Guo, F.; Xu, C.; Li, S.; Li, W.; Block, T.M.; Chang, J.; et al. Alpha-Interferon Suppresses Hepadnavirus Transcription by Altering Epigenetic Modification of cccDNA Minichromosomes. PLoS Pathog. 2013, 9, e1003613. [Google Scholar] [CrossRef]
- Belloni, L.; Pollicino, T.; De Nicola, F.; Guerrieri, F.; Raffa, G.; Fanciulli, M.; Raimondo, G.; Levrero, M. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc. Natl. Acad. Sci. USA 2009, 106, 19975–19979. [Google Scholar] [CrossRef]
- Zhang, T.; Xie, N.; He, W.; Liu, R.; Lei, Y.; Chen, Y.; Tang, H.; Liu, B.; Huang, C.; Wei, Y. An integrated proteomics and bioinformatics analyses of hepatitis B virus X interacting proteins and identification of a novel interactor apoA-I. J. Proteom. 2013, 84, 92–105. [Google Scholar] [CrossRef]
- Hu, J.; Seeger, C. Hsp90 is required for the activity of a hepatitis B virus reverse transcriptase. Proc. Natl. Acad. Sci. USA 1996, 93, 1060–1064. [Google Scholar] [CrossRef]
- Hu, J.; Toft, D.O.; Seeger, C. Hepadnavirus assembly and reverse transcription require a multi-component chaperone complex which is incorporated into nucleocapsids. EMBO J. 1997, 16, 59–68. [Google Scholar] [CrossRef]
- Hu, J.; Flores, D.; Toft, D.; Wang, X.; Nguyen, D. Requirement of heat shock protein 90 for human hepatitis B virus reverse transcriptase function. J. Virol. 2004, 78, 13122–13131. [Google Scholar] [CrossRef]
- Tsukamoto, Y.; Ikeda, S.; Uwai, K.; Taguchi, R.; Chayama, K.; Sakaguchi, T.; Narita, R.; Yao, W.L.; Takeuchi, F.; Otakaki, Y.; et al. Rosmarinic acid is a novel inhibitor for Hepatitis B virus replication targeting viral epsilon RNA-polymerase interaction. PLoS ONE 2018, 13, e0197664. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Hu, J. Inhibition of Hepadnavirus Reverse Transcriptase-ɛ RNA Interaction by Porphyrin Compounds. J. Virol. 2008, 82, 2305–2312. [Google Scholar] [CrossRef] [PubMed]
- Jo, E.; Ryu, D.K.; König, A.; Park, S.; Cho, Y.; Park, S.H.; Kim, T.H.; Yoon, S.K.; Ryu, W.S.; Cechetto, J.; et al. Identification and characterization of a novel hepatitis B virus pregenomic RNA encapsidation inhibitor. Antiviral Res. 2020, 175, 104709. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Beck, J.; Nassal, M.; Hu, K.H. A SELEX-Screened Aptamer of Human Hepatitis B Virus RNA Encapsidation Signal Suppresses Viral Replication. PLoS ONE 2011, 6, e27862. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Boregowda, R.; Spratt, T.E.; Hu, J. In vitro epsilon RNA-dependent protein priming activity of human hepatitis B virus polymerase. J. Virol. 2012, 86, 5134–5150. [Google Scholar] [CrossRef]
- Wang, X.; Hu, J. Distinct Requirement for Two Stages of Protein-Primed Initiation of Reverse Transcription in Hepadnaviruses. J. Virol. 2002, 76, 5857. [Google Scholar] [CrossRef]
- Jones, S.A.; Hu, J. Protein-primed terminal transferase activity of hepatitis B virus polymerase. J. Virol. 2013, 87, 2563–2576. [Google Scholar] [CrossRef]
- Tchesnokov, E.P.; Obikhod, A.; Schinazi, R.F.; Götte, M. Delayed Chain Termination Protects the Anti-hepatitis B Virus Drug Entecavir from Excision by HIV-1 Reverse Transcriptase. J. Biol. Chem. 2008, 283, 34218–34228. [Google Scholar] [CrossRef]
- Langley, D.R.; Walsh, A.W.; Baldick, C.J.; Eggers, B.J.; Rose, R.E.; Levine, S.M.; Kapur, A.J.; Colonno, R.J.; Tenney, D.J. Inhibition of hepatitis B virus polymerase by entecavir. J. Virol. 2007, 81, 3992–4001. [Google Scholar] [CrossRef]
- Boregowda, R.K.; Adams, C.; Hu, J. TP-RT Domain Interactions of Duck Hepatitis B Virus Reverse Transcriptase in cis and in trans during Protein-Primed Initiation of DNA Synthesis In Vitro. J. Virol. 2012, 86, 6522–6536. [Google Scholar] [CrossRef]
- Tavis, J.E.; Cheng, X.; Hu, Y.; Totten, M.; Cao, F.; Michailidis, E.; Aurora, R.; Meyers, M.J.; Jacobsen, E.J.; Parniak, M.A.; et al. The Hepatitis B Virus Ribonuclease H Is Sensitive to Inhibitors of the Human Immunodeficiency Virus Ribonuclease H and Integrase Enzymes. PLoS Pathog. 2013, 9, e1003125. [Google Scholar] [CrossRef]
- Hu, Y.; Cheng, X.; Cao, F.; Huang, A.; Tavis, J.E. β-Thujaplicinol inhibits hepatitis B virus replication by blocking the viral ribonuclease H activity. Antivir. Res. 2013, 99, 221–229. [Google Scholar] [CrossRef]
- Villa, J.A.; Pike, D.P.; Patel, K.B.; Lomonosova, E.; Lu, G.; Abdulqader, R.; Tavis, J.E. Purification and enzymatic characterization of the hepatitis B virus ribonuclease H, a new target for antiviral inhibitors. Antivir. Res. 2016, 132, 186–195. [Google Scholar] [CrossRef]
- Edwards, T.C.; Mani, N.; Dorsey, B.; Kakarla, R.; Rijnbrand, R.; Sofia, M.J.; Tavis, J.E. Inhibition of HBV replication by N-hydroxyisoquinolinedione and N-hydroxypyridinedione ribonuclease H inhibitors. Antivir. Res. 2019, 164, 70–80. [Google Scholar] [CrossRef]
- Edwards, T.C.; Lomonosova, E.; Patel, J.A.; Li, Q.; Villa, J.A.; Gupta, A.K.; Morrison, L.A.; Bailly, F.; Cotelle, P.; Giannakopoulou, E.; et al. Inhibition of hepatitis B virus replication by N-hydroxyisoquinolinediones and related polyoxygenated heterocycles. Antivir. Res. 2017, 143, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Huber, A.D.; Michailidis, E.; Tang, J.; Puray-Chavez, M.N.; Boftsi, M.; Wolf, J.J.; Boschert, K.N.; Sheridan, M.A.; Leslie, M.D.; Kirby, K.A.; et al. 3-hydroxypyrimidine-2,4-diones as novel hepatitis b virus antivirals targeting the viral ribonuclease H. Antimicrob. Agents Chemother. 2017, 61, e00245-17. [Google Scholar] [CrossRef]
- Lomonosova, E.; Zlotnick, A.; Tavis, J.E. Synergistic interactions between hepatitis B virus RNase H antagonists and other inhibitors. Antimicrob. Agents Chemother. 2017, 61, e02441-16. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Lomonosova, E.; Cheng, X.; Moran, E.A.; Meyers, M.J.; Le Grice, S.F.J.; Thomas, C.J.; Jiang, J.K.; Meck, C.; Hirsch, D.R.; et al. Hydroxylated tropolones inhibit hepatitis B virus replication by blocking viral ribonuclease H activity. Antimicrob. Agents Chemother. 2015, 59, 1070–1079. [Google Scholar] [CrossRef]
- Lu, G.; Villa, J.A.; Donlin, M.J.; Edwards, T.C.; Cheng, X.; Heier, R.F.; Meyers, M.J.; Tavis, J.E. Hepatitis B virus genetic diversity has minimal impact on sensitivity of the viral ribonuclease H to inhibitors. Antivir. Res. 2016, 135, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, C. HBV and the immune response. Liver Int. 2015, 35 (Suppl. S1), 121–128. [Google Scholar] [CrossRef]
- Rehermann, B. Pathogenesis of chronic viral hepatitis: Differential roles of T cells and NK cells. Nat. Med. 2013, 19, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Protzer, U.; Maini, M.K.; Knolle, P.A. Living in the liver: Hepatic infections. Nat. Rev. Immunol. 2012, 12, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, L.G.; Chisari, F.V. Immunobiology and pathogenesis of viral hepatitis. Annu. Rev. Pathol. 2006, 1, 23–61. [Google Scholar] [CrossRef] [PubMed]
- Rehermann, B.; Ferrari, C.; Pasquinelli, C.; Chisari, F.V. The hepatitis B virus persists for decades after patients’ recovery from acute viral hepatitis despite active maintenance of a cytotoxic T-lymphocyte response. Nat. Med. 1996, 2, 1104–1108. [Google Scholar] [CrossRef] [PubMed]
- Moraleda, G.; Saputelli, J.; Aldrich, C.E.; Averett, D.; Condreay, L.; Mason, W.S. Lack of effect of antiviral therapy in nondividing hepatocyte cultures on the closed circular DNA of woodchuck hepatitis virus. J. Virol. 1997, 71, 9392–9399. [Google Scholar] [CrossRef]
- Chisari, F.V.; Isogawa, M.; Wieland, S.F. Pathogenesis of Hepatitis B Virus Infection. Pathol. Biol. 2010, 58, 258–266. [Google Scholar] [CrossRef]
- Mason, W.S.; Xu, C.; Low, H.C.; Saputelli, J.; Aldrich, C.E.; Scougall, C.; Grosse, A.; Colonno, R.; Litwin, S.; Jilbert, A.R. The Amount of Hepatocyte Turnover That Occurred during Resolution of Transient Hepadnavirus Infections Was Lower When Virus Replication Was Inhibited with Entecavir. J. Virol. 2009, 83, 1778–1789. [Google Scholar] [CrossRef]
- Murray, J.M.; Goyal, A. In silico single cell dynamics of hepatitis B virus infection and clearance. J. Theor. Biol. 2015, 366, 91–102. [Google Scholar] [CrossRef]
- Murray, J.M.; Wieland, S.F.; Purcell, R.H.; Chisari, F.V. Dynamics of hepatitis B virus clearance in chimpanzees. Proc. Natl. Acad. Sci. USA 2005, 102, 17780–17785. [Google Scholar] [CrossRef]
- Lanford, R.E.; Guerra, B.; Chavez, D.; Giavedoni, L.; Hodara, V.L.; Brasky, K.M.; Fosdick, A.; Frey, C.R.; Zheng, J.; Wolfgang, G.; et al. GS-9620, an oral agonist of Toll-like receptor-7, induces prolonged suppression of hepatitis B virus in chronically infected chimpanzees. Gastroenterology 2013, 144, 1508–1517. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Kim, Y.H.; Kim, J.S.; Kim, H. Genome engineering in human cells. Methods Enzymol. 2014, 546, 93–118. [Google Scholar] [CrossRef] [PubMed]
- Weber, N.D.; Stone, D.; Sedlak, R.H.; De Silva Feelixge, H.S.; Roychoudhury, P.; Schiffer, J.T.; Aubert, M.; Jerome, K.R. AAV-Mediated Delivery of Zinc Finger Nucleases Targeting Hepatitis B Virus Inhibits Active Replication. PLoS ONE 2014, 9, e97579. [Google Scholar] [CrossRef] [PubMed]
- Cradick, T.J.; Keck, K.; Bradshaw, S.; Jamieson, A.C.; McCaffrey, A.P. Zinc-finger nucleases as a novel therapeutic strategy for targeting hepatitis B virus DNAs. Mol. Ther. 2010, 18, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Bloom, K.; Ely, A.; Mussolino, C.; Cathomen, T.; Arbuthnot, P. Inactivation of hepatitis B virus replication in cultured cells and in vivo with engineered transcription activator-like effector nucleases. Mol. Ther. 2013, 21, 1889–1897. [Google Scholar] [CrossRef]
- Seeger, C.; Sohn, J.A. Targeting Hepatitis B Virus With CRISPR/Cas9. Mol. Ther. Nucleic Acids 2014, 3, e216. [Google Scholar] [CrossRef]
- Hu, J.; Boyer, M. Hepatitis B Virus Reverse Transcriptase and ε RNA Sequences Required for Specific Interaction In Vitro. J. Virol. 2006, 80, 2141–2150. [Google Scholar] [CrossRef]
- Feng, H.; Chen, P.; Zhao, F.; Nassal, M.; Hu, K. Evidence for Multiple Distinct Interactions between Hepatitis B Virus P Protein and Its Cognate RNA Encapsidation Signal during Initiation of Reverse Transcription. PLoS ONE 2013, 8, e72798. [Google Scholar] [CrossRef]
- Lok, A.S.F.; Akarca, U.; Greene, S. Mutations in the pre-core region of hepatitis B virus serve to enhance the stability of the secondary structure of the pre-genome encapsidation signal. Proc. Natl. Acad. Sci. USA 1994, 91, 4077–4081. [Google Scholar] [CrossRef]
- Laskus, T.; Rakela, J.; Persing, D.H. The stem-loop structure of the cis-encapsidation signal is highly conserved in naturally occurring hepatitis B virus variants. Virology 1994, 200, 809–812. [Google Scholar] [CrossRef]
- Flodell, S.; Schleucher, J.; Cromsigt, J.; Ippel, H.; Kidd-Ljunggren, K.; Wijmenga, S. The apical stem–loop of the hepatitis B virus encapsidation signal folds into a stable tri–loop with two underlying pyrimidine bulges. Nucleic Acids Res. 2002, 30, 4803–4811. [Google Scholar] [CrossRef] [PubMed]
- Flodell, S.; Petersen, M.; Girard, F.; Zdunek, J.; Kidd-Ljunggren, K.; Schleucher, J.; Wijmenga, S. Solution structure of the apical stem-loop of the human hepatitis B virus encapsidation signal. Nucleic Acids Res. 2006, 34, 4449–4457. [Google Scholar] [CrossRef] [PubMed]
- Petzold, K.; Duchardt, E.; Flodell, S.; Larsson, G.; Kidd-Ljunggren, K.; Wijmenga, S.; Schleucher, J. Conserved nucleotides in an RNA essential for hepatitis B virus replication show distinct mobility patterns. Nucleic Acids Res. 2007, 35, 6854–6861. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Xiong, X.; Yang, H.; Westland, C.E.; Gibbs, C.S.; Sarafianos, S.G.; Arnold, E. Molecular modeling and biochemical characterization reveal the mechanism of hepatitis B virus polymerase resistance to lamivudine (3TC) and emtricitabine (FTC). J. Virol. 2001, 75, 4771–4779. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.I.; Deslauriers, M.; Webster Andrews, C.; Tipples, G.A.; Walters, K.A.; Tyrrell, D.L.J.; Brown, N.; Condreay, L.D. Identification and characterization of mutations in hepatitis B virus resistant to lamivudine. Lamivudine Clinical Investigation Group. Hepatology 1998, 27, 1670–1677. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.; Vogel, M.; Nassal, M. dNTP versus NTP discrimination by phenylalanine 451 in duck hepatitis B virus P protein indicates a common structure of the dNTP-binding pocket with other reverse transcriptases. Nucleic Acids Res. 2002, 30, 1679–1687. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Anselmo, D. In vitro reconstitution of a functional duck hepatitis B virus reverse transcriptase: Posttranslational activation by Hsp90. J. Virol. 2000, 74, 11447–11455. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.; Nassal, M. In vitro reconstitution of epsilon-dependent duck hepatitis B virus replication initiation. Methods Mol. Med. 2004, 95, 315–325. [Google Scholar] [CrossRef]
- Beck, J.; Nassal, M. Reconstitution of a functional duck hepatitis B virus replication initiation complex from separate reverse transcriptase domains expressed in Escherichia coli. J. Virol. 2001, 75, 7410–7419. [Google Scholar] [CrossRef]
- Buhlig, T.S.; Bowersox, A.F.; Braun, D.L.; Owsley, D.N.; James, K.D.; Aranda, A.J.; Kendrick, C.D.; Skalka, N.A.; Clark, D.N. Molecular, Evolutionary, and Structural Analysis of the Terminal Protein Domain of Hepatitis B Virus Polymerase, a Potential Drug Target. Viruses 2020, 12, 570. [Google Scholar] [CrossRef]
- Tajwar, R.; Bradley, D.P.; Ponzar, N.L.; Tavis, J.E.; Tavis, J. Predicted structure of the hepatitis B virus polymerase reveals an ancient conserved protein fold. Protein Sci. 2022, 31, e4421. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Wang, H.; Ryu, W.-S. Incorporation of eukaryotic translation initiation factor eIF4E into viral nucleocapsids via interaction with hepatitis B virus polymerase. J. Virol. 2010, 84, 52–58. [Google Scholar] [CrossRef]
- Nguyen, D.H.; Hu, J. Reverse transcriptase- and RNA packaging signal-dependent incorporation of APOBEC3G into hepatitis B virus nucleocapsids. J. Virol. 2008, 82, 6852–6861. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Sun, T.; Li, G.; Pan, W.; Wang, K.; Dai, J. DEAD-box helicase DDX25 is a negative regulator of type I interferon pathway and facilitates RNA virus infection. Front. Cell. Infect. Microbiol. 2017, 7, 356. [Google Scholar] [CrossRef]
- Wang, H.; Kim, S.; Ryu, W.-S. DDX3 DEAD-Box RNA helicase inhibits hepatitis B virus reverse transcription by incorporation into nucleocapsids. J. Virol. 2009, 83, 5815–5824. [Google Scholar] [CrossRef]
- Chen, J.; Wu, M.; Zhang, X.; Zhang, W.; Zhang, Z.; Chen, L.; He, J.; Zheng, Y.; Chen, C.; Wang, F.; et al. Hepatitis B virus polymerase impairs interferon-α-induced STA T activation through inhibition of importin-α5 and protein kinase C-δ. Hepatology 2013, 57, 470–482. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Clark, D.N.; Cao, F.; Tavis, J.E.; Hu, J. Comparative analysis of hepatitis B virus polymerase sequences required for viral RNA binding, RNA packaging, and protein priming. J. Virol. 2014, 88, 1564–1572. [Google Scholar] [CrossRef] [PubMed]
- Olenginski, L.T.; Kasprzak, W.K.; Bergonzo, C.; Shapiro, B.A.; Dayie, T.K. Conformational dynamics of the hepatitis B virus pre-genomic RNA on multiple time scales: Implications for viral replication. J. Mol. Biol. 2022, 434, 167633. [Google Scholar] [CrossRef]
- Schwieters, C.D.; Bermejo, G.A.; Clore, G.M. Xplor-NIH for molecular structure determination from NMR and other data sources. Protein Sci. 2018, 27, 26–40. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Moxon, S.; Marshall, M.; Khanna, A.; Eddy, S.R.; Bateman, A. Rfam: Annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005, 33, D121–D124. [Google Scholar] [CrossRef]
- Kalvari, I.; Nawrocki, E.P.; Ontiveros-Palacios, N.; Argasinska, J.; Lamkiewicz, K.; Marz, M.; Griffiths-Jones, S.; Toffano-Nioche, C.; Gautheret, D.; Weinberg, Z.; et al. Rfam 14: Expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 2021, 49, D192–D200. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S.; Bateman, A.; Marshall, M.; Khanna, A.; Eddy, S.R. Rfam: An RNA family database. Nucleic Acids Res. 2003, 31, 439–441. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.; Nassal, M. Sequence- and structure-specific determinants in the interaction between the RNA encapsidation signal and reverse transcriptase of avian hepatitis B viruses. J. Virol. 1997, 71, 4971–4980. [Google Scholar] [CrossRef] [PubMed]
- Hargrove, A.E. Small molecule–RNA targeting: Starting with the fundamentals. Chem. Commun. 2020, 56, 14744–14756. [Google Scholar] [CrossRef]
- Falese, J.P.; Donlic, A.; Hargrove, A.E. Targeting RNA with small molecules: From fundamental principles towards the clinic. Chem. Soc. Rev. 2021, 50, 2224–2243. [Google Scholar] [CrossRef] [PubMed]
- Shortridge, M.D.; Varani, G. Structure based approaches for targeting non-coding RNAs with small molecules. Curr. Opin. Struct. Biol. 2015, 30, 79–88. [Google Scholar] [CrossRef]
- Disney, M.D.; Yildirim, I.; Childs-Disney, J.L. Methods to enable the design of bioactive small molecules targeting RNA. Org. Biomol. Chem. 2014, 12, 1029–1039. [Google Scholar] [CrossRef]
- Naryshkin, N.A.; Weetall, M.; Dakka, A.; Narasimhan, J.; Zhao, X.; Feng, Z.; Ling, K.K.Y.; Karp, G.M.; Qi, H.; Woll, M.G.; et al. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science 2014, 345, 688–693. [Google Scholar] [CrossRef]
- Stelzer, A.C.; Frank, A.T.; Kratz, J.D.; Swanson, M.D.; Gonzalez-Hernandez, M.J.; Lee, J.; Andricioaei, I.; Markovitz, D.M.; Al-Hashimi, H.M. Discovery of selective bioactive small molecules by targeting an RNA dynamic ensemble. Nat. Chem. Biol. 2011, 7, 553–559. [Google Scholar] [CrossRef]
- Abulwerdi, F.A.; Shortridge, M.D.; Sztuba-Solinska, J.; Wilson, R.; Le Grice, S.F.J.; Varani, G.; Schneekloth, J.S. Development of Small Molecules with a Noncanonical Binding Mode to HIV-1 Trans Activation Response (TAR) RNA. J. Med. Chem. 2016, 59, 11148–11160. [Google Scholar] [CrossRef]
- Connelly, C.M.; Boer, R.E.; Moon, M.H.; Gareiss, P.; Schneekloth, J.S. Discovery of Inhibitors of MicroRNA-21 Processing Using Small Molecule Microarrays. ACS Chem. Biol. 2017, 12, 435–443. [Google Scholar] [CrossRef]
- Connelly, C.M.; Numata, T.; Boer, R.E.; Moon, M.H.; Sinniah, R.S.; Barchi, J.J.; Ferré-D’Amaré, A.R.; Schneekloth, J.S. Synthetic ligands for PreQ1 riboswitches provide structural and mechanistic insights into targeting RNA tertiary structure. Nat. Commun. 2019, 10, 1501. [Google Scholar] [CrossRef] [PubMed]
- Abulwerdi, F.A.; Xu, W.; Ageeli, A.A.; Yonkunas, M.J.; Arun, G.; Nam, H.; Schneekloth, J.S.; Dayie, T.K.; Spector, D.; Baird, N.; et al. Selective Small-Molecule Targeting of a Triple Helix Encoded by the Long Noncoding RNA, MALAT1. ACS Chem. Biol. 2019, 14, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, K.M.; Saunders, L.B.; Simmons, J.K.; Leon, E.; Calabrese, D.R.; Zhang, S.; Michalowski, A.; Gareiss, P.; Mock, B.A.; Schneekloth, J.S. Small Molecule Microarrays Enable the Identification of a Selective, Quadruplex-Binding Inhibitor of MYC Expression. ACS Chem. Biol. 2016, 11, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, D.R.; Chen, X.; Leon, E.C.; Gaikwad, S.M.; Phyo, Z.; Hewitt, W.M.; Alden, S.; Hilimire, T.A.; He, F.; Michalowski, A.M.; et al. Chemical and structural studies provide a mechanistic basis for recognition of the MYC G-quadruplex. Nat. Commun. 2018, 9, 4229. [Google Scholar] [CrossRef]
- Valentovic, M. Gemfibrozil. In xPharm: The Comprehensive Pharmacology Reference; Elsevier: Amsterdam, The Netherlands, 2007; pp. 1–5. [Google Scholar] [CrossRef]
- Maximov, P.Y.; Lee, T.M.; Craig Jordan, V. The discovery and development of selective estrogen receptor modulators (SERMs) for clinical practice. Curr. Clin. Pharmacol. 2013, 8, 135–155. [Google Scholar] [CrossRef]
- Bak, E.; Miller, J.T.; Noronha, A.; Tavis, J.; Gallicchio, E.; Murelli, R.P.; Le Grice, S.F.J. 3,7-Dihydroxytropolones Inhibit Initiation of Hepatitis B Virus Minus-Strand DNA Synthesis. Molecules 2020, 25, 4434. [Google Scholar] [CrossRef]
- Olenginski, L.T.; Kasprzak, W.K.; Attionu, S.K.; Shapiro, B.A.; Dayie, T.K. Virtual Screening of Hepatitis B Virus Pre-Genomic RNA as a Novel Therapeutic Target. Molecules 2023, 28, 1803. [Google Scholar] [CrossRef]
- Carlson, H.A.; Masukawa, K.M.; Rubins, K.; Bushman, F.D.; Jorgensen, W.L.; Lins, R.D.; Briggs, J.M.; McCammon, J.A. Developing a dynamic pharmacophore model for HIV-1 integrase. J. Med. Chem. 2000, 43, 2100–2114. [Google Scholar] [CrossRef]
- Lin, J.H.; Perryman, A.L.; Schames, J.R.; McCammon, J.A. Computational drug design accommodating receptor flexibility: The relaxed complex scheme. J. Am. Chem. Soc. 2002, 124, 5632–5633. [Google Scholar] [CrossRef]
- Knegtel, R.M.A.; Kuntz, I.D.; Oshiro, C.M. Molecular docking to ensembles of protein structures. J. Mol. Biol. 1997, 266, 424–440. [Google Scholar] [CrossRef] [PubMed]
- Ganser, L.R.; Lee, J.; Rangadurai, A.; Merriman, D.K.; Kelly, M.L.; Kansal, A.D.; Sathyamoorthy, B.; Al-Hashimi, H.M. High-performance virtual screening by targeting a high-resolution RNA dynamic ensemble. Nat. Struct. Mol. Biol. 2018, 25, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Tóth, G.; Gardai, S.J.; Zago, W.; Bertoncini, C.W.; Cremades, N.; Roy, S.L.; Tambe, M.A.; Rochet, J.C.; Galvagnion, C.; Skibinski, G.; et al. Targeting the intrinsically disordered structural ensemble of α-synuclein by small molecules as a potential therapeutic strategy for Parkinson’s disease. PLoS ONE 2014, 9, e87133. [Google Scholar] [CrossRef]
- Fischer, M.; Coleman, R.G.; Fraser, J.S.; Shoichet, B.K. Incorporation of protein flexibility and conformational energy penalties in docking screens to improve ligand discovery. Nat. Chem. 2014, 6, 575–583. [Google Scholar] [CrossRef]
- Salmon, L.; Yang, S.; Al-Hashimi, H.M. Advances in the determination of nucleic acid conformational ensembles. Annu. Rev. Phys. Chem. 2014, 65, 293–316. [Google Scholar] [CrossRef] [PubMed]
- Salmon, L.; Bascom, G.; Andricioaei, I.; Al-Hashimi, H.M. A general method for constructing atomic-resolution RNA ensembles using NMR residual dipolar couplings: The basis for interhelical motions revealed. J. Am. Chem. Soc. 2013, 135, 5457–5466. [Google Scholar] [CrossRef]
- Frank, A.T.; Stelzer, A.C.; Al-Hashimi, H.M.; Andricioaei, I. Constructing RNA dynamical ensembles by combining MD and motionally decoupled NMR RDCs: New insights into RNA dynamics and adaptive ligand recognition. Nucleic Acids Res. 2009, 37, 3670–3679. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. ZINC-A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15–Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olenginski, L.T.; Attionu, S.K.; Henninger, E.N.; LeBlanc, R.M.; Longhini, A.P.; Dayie, T.K. Hepatitis B Virus Epsilon (ε) RNA Element: Dynamic Regulator of Viral Replication and Attractive Therapeutic Target. Viruses 2023, 15, 1913. https://doi.org/10.3390/v15091913
Olenginski LT, Attionu SK, Henninger EN, LeBlanc RM, Longhini AP, Dayie TK. Hepatitis B Virus Epsilon (ε) RNA Element: Dynamic Regulator of Viral Replication and Attractive Therapeutic Target. Viruses. 2023; 15(9):1913. https://doi.org/10.3390/v15091913
Chicago/Turabian StyleOlenginski, Lukasz T., Solomon K. Attionu, Erica N. Henninger, Regan M. LeBlanc, Andrew P. Longhini, and Theodore K. Dayie. 2023. "Hepatitis B Virus Epsilon (ε) RNA Element: Dynamic Regulator of Viral Replication and Attractive Therapeutic Target" Viruses 15, no. 9: 1913. https://doi.org/10.3390/v15091913