
Mmp12 Is Translationally Regulated in Macrophages during the Course of Inflammation
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Efferocytosis Alters Inflammatory Responses in Macrophages
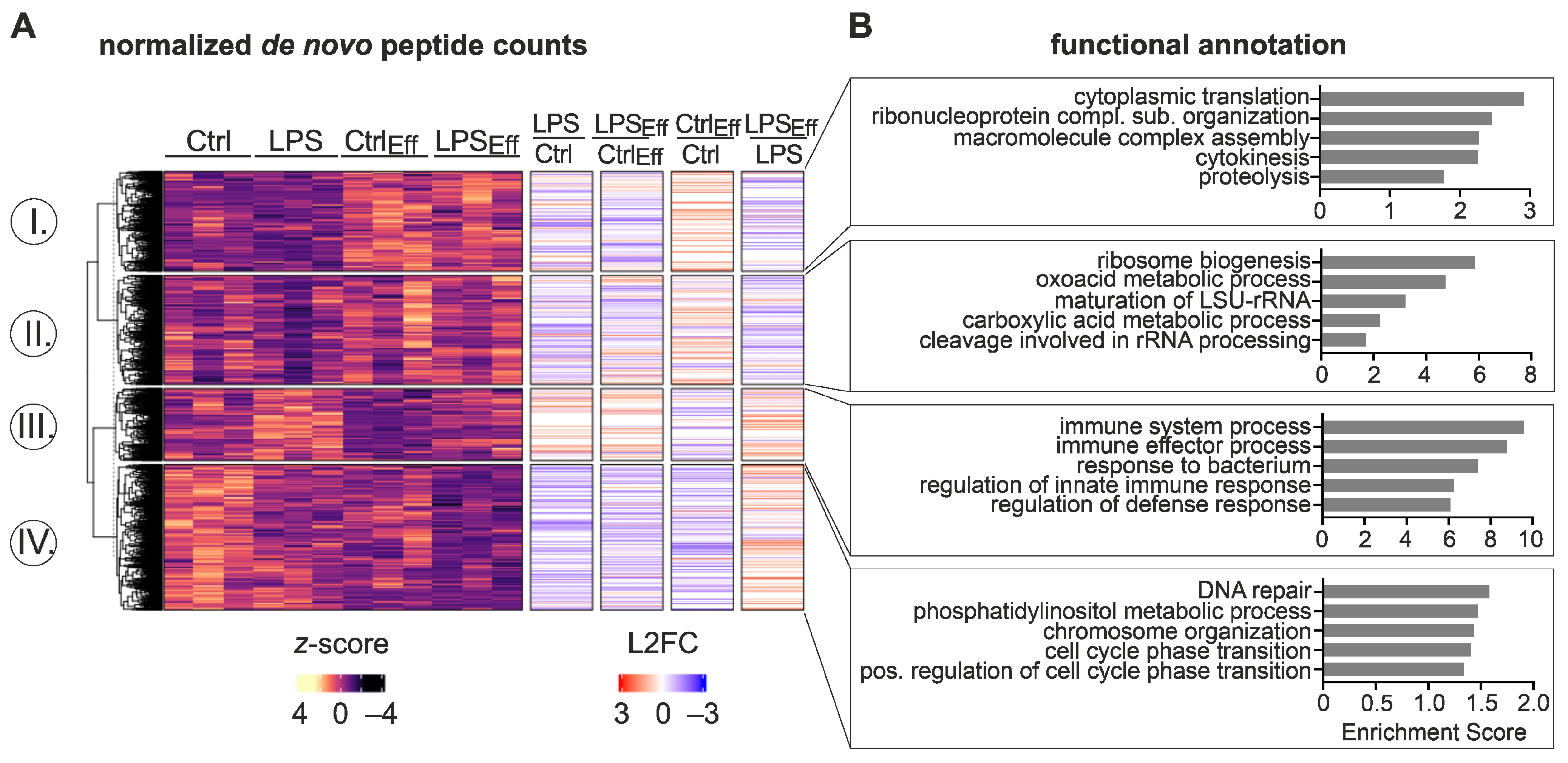
2.2. Influence of Efferocytosis on Inflammatory mRNA Expression and Translation
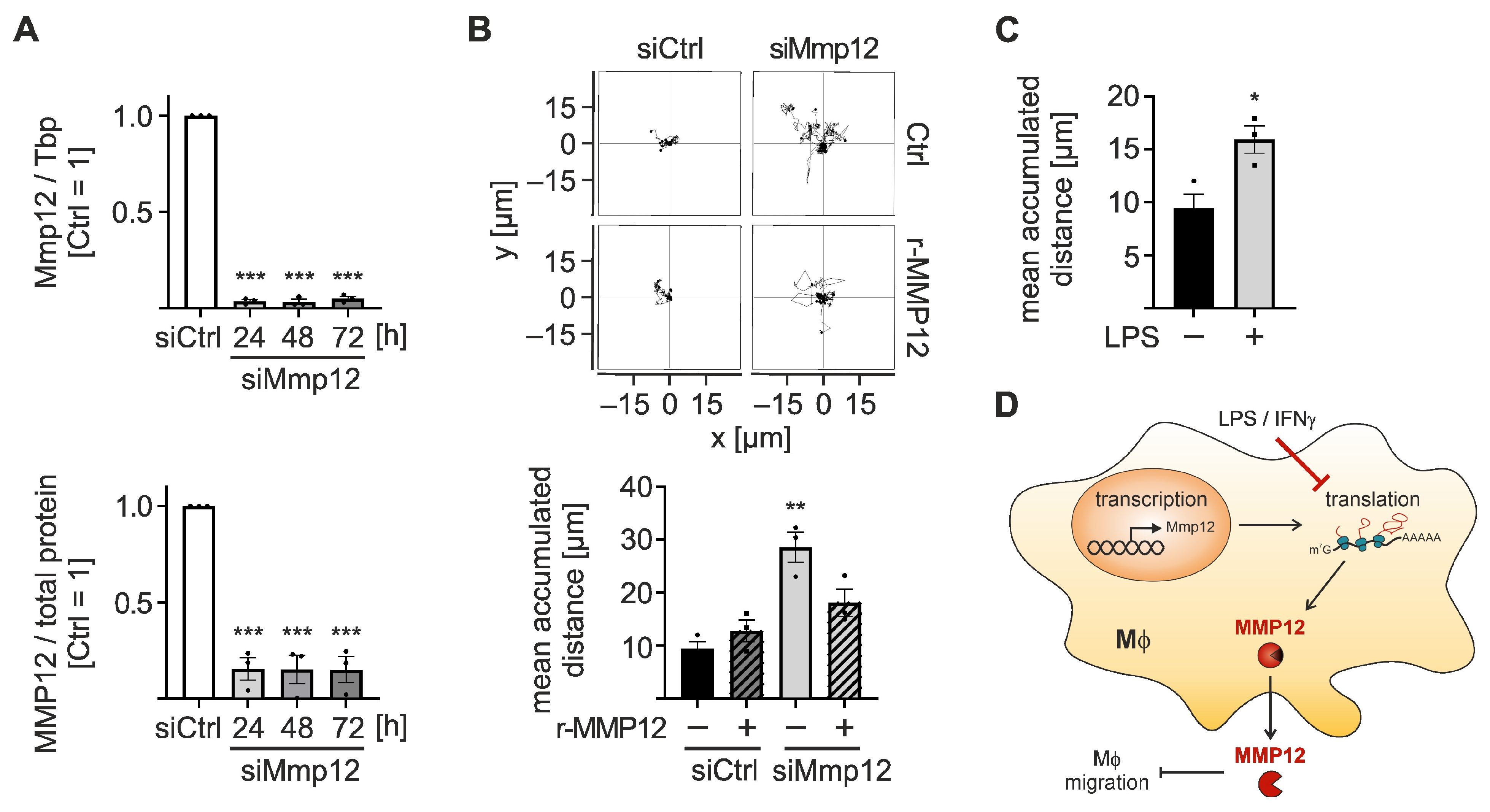
2.3. Characterization of Matrix Metallopeptidase 12 (Mmp12) Regulation throughout the Course of Inflammation
2.4. MMP12 Suppresses Migration of Macrophages
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Chemicals
5.2. Cell Culture
5.3. Efferocytosis Model
5.4. Viability Assay
5.5. Efferocytosis Assays
5.6. RNA Isolation, Reverse Transcription, and Quantitative Polymerase Chain Reaction (RT-qPCR)
5.7. Western Blot Analysis and ELISA
5.8. Polysomal Fractionation
5.9. RNA Sequencing
5.10. De Novo Proteomics (Multiplexed Enhanced Protein Dynamics (mePROD) Proteomics)
5.11. Migration Assay
5.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fioranelli, M.; Roccia, M.G.; Flavin, D.; Cota, L. Regulation of Inflammatory Reaction in Health and Disease. Int. J. Mol. Sci. 2021, 22, 5277. [Google Scholar] [CrossRef]
- Dalli, J.; Serhan, C.N. Pro-Resolving Mediators in Regulating and Conferring Macrophage Function. Front. Immunol. 2017, 8, 1400. [Google Scholar] [CrossRef]
- Fullerton, J.N.; Gilroy, D.W. Resolution of inflammation: A new therapeutic frontier. Nat. Rev. Drug Discov. 2016, 15, 551–567. [Google Scholar] [CrossRef]
- Savill, J.S.; Wyllie, A.H.; Henson, J.E.; Walport, M.J.; Henson, P.M.; Haslett, C. Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. J. Clin. Investig. 1989, 83, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Gautier, E.L.; Ivanov, S.; Lesnik, P.; Randolph, G.J. Local apoptosis mediates clearance of macrophages from resolving inflammation in mice. Blood 2013, 122, 2714–2722. [Google Scholar] [CrossRef]
- Doran, A.C.; Yurdagul, A.; Tabas, I. Efferocytosis in health and disease. Nat. Rev. Immunol. 2020, 20, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Razi, S.; Yaghmoorian Khojini, J.; Kargarijam, F.; Panahi, S.; Tahershamsi, Z.; Tajbakhsh, A.; Gheibihayat, S.M. Macrophage efferocytosis in health and disease. Cell Biochem. Funct. 2023, 41, 152–165. [Google Scholar] [CrossRef]
- Elliott, M.R.; Koster, K.M.; Murphy, P.S. Efferocytosis Signaling in the Regulation of Macrophage Inflammatory Responses. J. Immunol. 2017, 198, 1387–1394. [Google Scholar] [CrossRef]
- Fadok, V.A.; Bratton, D.L.; Konowal, A.; Freed, P.W.; Westcott, J.Y.; Henson, P.M. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Investig. 1998, 101, 890–898. [Google Scholar] [CrossRef]
- Schilperoort, M.; Ngai, D.; Sukka, S.R.; Avrampou, K.; Shi, H.; Tabas, I. The role of efferocytosis-fueled macrophage metabolism in the resolution of inflammation. Immunol. Rev. 2023, 319, 65–80. [Google Scholar] [CrossRef]
- Collins, G.; Souza Carvalho, J.; de Gilroy, D.W. The translation potential of harnessing the resolution of inflammation. J. Allergy Clin. Immunol. 2023, 152, 356–358. [Google Scholar] [CrossRef] [PubMed]
- Saas, P.; Vetter, M.; Maraux, M.; Bonnefoy, F.; Perruche, S. Resolution therapy: Harnessing efferocytic macrophages to trigger the resolution of inflammation. Front. Immunol. 2022, 13, 1021413. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ding, W.; Zhao, M.; Liu, J.; Xu, Y.; Wan, J.; Wang, M. Mechanisms of efferocytosis in determining inflammation resolution: Therapeutic potential and the association with cardiovascular disease. Br. J. Pharmacol. 2022, 179, 5151–5171. [Google Scholar] [CrossRef] [PubMed]
- Das, A.S.; Basu, A.; Kumar, R.; Borah, P.K.; Bakshi, S.; Sharma, M.; Duary, R.K.; Ray, P.S.; Mukhopadhyay, R. Post-transcriptional regulation of C-C motif chemokine ligand 2 expression by ribosomal protein L22 during LPS-mediated inflammation. FEBS J. 2020, 287, 3794–3813. [Google Scholar] [CrossRef]
- Naqvi, R.A.; Gupta, M.; George, A.; Naqvi, A.R. MicroRNAs in shaping the resolution phase of inflammation. Semin. Cell Dev. Biol. 2022, 124, 48–62. [Google Scholar] [CrossRef]
- Rappl, P.; Brüne, B.; Schmid, T. Role of Tristetraprolin in the Resolution of Inflammation. Biology 2021, 10, 66. [Google Scholar] [CrossRef] [PubMed]
- Piccirillo, C.A.; Bjur, E.; Topisirovic, I.; Sonenberg, N.; Larsson, O. Translational control of immune responses: From transcripts to translatomes. Nat. Immunol. 2014, 15, 503–511. [Google Scholar] [CrossRef]
- Bosurgi, L.; Cao, Y.G.; Cabeza-Cabrerizo, M.; Tucci, A.; Hughes, L.D.; Kong, Y.; Weinstein, J.S.; Licona-Limon, P.; Schmid, E.T.; Pelorosso, F.; et al. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science 2017, 356, 1072–1076. [Google Scholar] [CrossRef]
- Rappl, P.; Rösser, S.; Maul, P.; Bauer, R.; Huard, A.; Schreiber, Y.; Thomas, D.; Geisslinger, G.; Jakobsson, P.-J.; Weigert, A.; et al. Inhibition of mPGES-1 attenuates efficient resolution of acute inflammation by enhancing CX3CL1 expression. Cell Death Dis. 2021, 12, 135. [Google Scholar] [CrossRef]
- Bartish, M.; Tong, D.; Pan, Y.; Wallerius, M.; Liu, H.; Ristau, J.; Souza Ferreira, S.; de Wallmann, T.; van Hoef, V.; Masvidal, L.; et al. MNK2 governs the macrophage antiinflammatory phenotype. Proc. Natl. Acad. Sci. USA 2020, 117, 27556–27565. [Google Scholar] [CrossRef]
- Knuth, A.-K.; Huard, A.; Naeem, Z.; Rappl, P.; Bauer, R.; Mota, A.C.; Schmid, T.; Fleming, I.; Brüne, B.; Fulda, S.; et al. Apoptotic Cells induce Proliferation of Peritoneal Macrophages. Int. J. Mol. Sci. 2021, 22, 2230. [Google Scholar] [CrossRef]
- Young, S.K.; Wek, R.C. Upstream Open Reading Frames Differentially Regulate Gene-specific Translation in the Integrated Stress Response. J. Biol. Chem. 2016, 291, 16927–16935. [Google Scholar] [CrossRef]
- Da Huang, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Klann, K.; Tascher, G.; Münch, C. Functional Translatome Proteomics Reveal Converging and Dose-Dependent Regulation by mTORC1 and eIF2α. Mol. Cell 2020, 77, 913–925.e4. [Google Scholar] [CrossRef] [PubMed]
- Dufour, A.; Bellac, C.L.; Eckhard, U.; Solis, N.; Klein, T.; Kappelhoff, R.; Fortelny, N.; Jobin, P.; Rozmus, J.; Mark, J.; et al. C-terminal truncation of IFN-γ inhibits proinflammatory macrophage responses and is deficient in autoimmune disease. Nat. Commun. 2018, 9, 2416. [Google Scholar] [CrossRef] [PubMed]
- Hautamaki, R.D.; Kobayashi, D.K.; Senior, R.M.; Shapiro, S.D. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science 1997, 277, 2002–2004. [Google Scholar] [CrossRef]
- Crick, F. Central dogma of molecular biology. Nature 1970, 227, 561–563. [Google Scholar] [CrossRef]
- Schott, J.; Reitter, S.; Philipp, J.; Haneke, K.; Schäfer, H.; Stoecklin, G. Translational regulation of specific mRNAs controls feedback inhibition and survival during macrophage activation. PLoS Genet. 2014, 10, e1004368. [Google Scholar] [CrossRef]
- Tebaldi, T.; Re, A.; Viero, G.; Pegoretti, I.; Passerini, A.; Blanzieri, E.; Quattrone, A. Widespread uncoupling between transcriptome and translatome variations after a stimulus in mammalian cells. BMC Genom. 2012, 13, 220. [Google Scholar] [CrossRef]
- Chaparro, V.; Leroux, L.-P.; Masvidal, L.; Lorent, J.; Graber, T.E.; Zimmermann, A.; Arango Duque, G.; Descoteaux, A.; Alain, T.; Larsson, O.; et al. Translational profiling of macrophages infected with Leishmania donovani identifies mTOR- and eIF4A-sensitive immune-related transcripts. PLoS Pathog. 2020, 16, e1008291. [Google Scholar] [CrossRef] [PubMed]
- Ashe, M.P.; de Long, S.K.; Sachs, A.B. Glucose depletion rapidly inhibits translation initiation in yeast. Mol. Biol. Cell 2000, 11, 833–848. [Google Scholar] [CrossRef]
- Zhang, P.; McGrath, B.C.; Reinert, J.; Olsen, D.S.; Lei, L.; Gill, S.; Wek, S.A.; Vattem, K.M.; Wek, R.C.; Kimball, S.R.; et al. The GCN2 eIF2alpha kinase is required for adaptation to amino acid deprivation in mice. Mol. Cell. Biol. 2002, 22, 6681–6688. [Google Scholar] [CrossRef] [PubMed]
- Vattem, K.M.; Wek, R.C. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11269–11274. [Google Scholar] [CrossRef] [PubMed]
- Han, C.Z.; Ravichandran, K.S. Metabolic connections during apoptotic cell engulfment. Cell 2011, 147, 1442–1445. [Google Scholar] [CrossRef]
- Ceppi, M.; Clavarino, G.; Gatti, E.; Schmidt, E.K.; de Gassart, A.; Blankenship, D.; Ogola, G.; Banchereau, J.; Chaussabel, D.; Pierre, P. Ribosomal protein mRNAs are translationally-regulated during human dendritic cells activation by LPS. Immunome Res. 2009, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Cockman, E.; Anderson, P.; Ivanov, P. TOP mRNPs: Molecular Mechanisms and Principles of Regulation. Biomolecules 2020, 10, 969. [Google Scholar] [CrossRef]
- Liu, B.; Qian, S.-B. Translational regulation in nutrigenomics. Adv. Nutr. 2011, 2, 511–519. [Google Scholar] [CrossRef]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef]
- Wu, L.; Fan, J.; Matsumoto, S.I.; Watanabe, T. Induction and regulation of matrix metalloproteinase-12 by cytokines and CD40 signaling in monocyte/macrophages. Biochem. Biophys. Res. Commun. 2000, 269, 808–815. [Google Scholar] [CrossRef]
- Jost, M.M.; Ninci, E.; Meder, B.; Kempf, C.; van Royen, N.; Hua, J.; Berger, B.; Hoefer, I.; Modolell, M.; Buschmann, I. Divergent effects of GM-CSF and TGFbeta1 on bone marrow-derived macrophage arginase-1 activity, MCP-1 expression, and matrix metalloproteinase-12: A potential role during arteriogenesis. FASEB J. 2003, 17, 2281–2283. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Shichiri, M.; Libby, P.; Lee, R.T.; Mitchell, R.N. Th2-predominant inflammation and blockade of IFN-γ signaling induce aneurysms in allografted aortas. J. Clin. Investig. 2004, 114, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, B.; Li, X.; Barik, S. Translation control: A multifaceted regulator of inflammatory response. J. Immunol. 2010, 184, 3311–3319. [Google Scholar] [CrossRef] [PubMed]
- Castrillo, A.; Joseph, S.B.; Marathe, C.; Mangelsdorf, D.J.; Tontonoz, P. Liver X receptor-dependent repression of matrix metalloproteinase-9 expression in macrophages. J. Biol. Chem. 2003, 278, 10443–10449. [Google Scholar] [CrossRef]
- Shapiro, S.D. Matrix metalloproteinase degradation of extracellular matrix: Biological consequences. Curr. Opin. Cell Biol. 1998, 10, 602–608. [Google Scholar] [CrossRef]
- Mouton, A.J.; Rivera Gonzalez, O.J.; Kaminski, A.R.; Moore, E.T.; Lindsey, M.L. Matrix metalloproteinase-12 as an endogenous resolution promoting factor following myocardial infarction. Pharmacol. Res. 2018, 137, 252–258. [Google Scholar] [CrossRef]
- Dean, R.A.; Cox, J.H.; Bellac, C.L.; Doucet, A.; Starr, A.E.; Overall, C.M. Macrophage-specific metalloelastase (MMP-12) truncates and inactivates ELR+ CXC chemokines and generates CCL2, -7, -8, and -13 antagonists: Potential role of the macrophage in terminating polymorphonuclear leukocyte influx. Blood 2008, 112, 3455–3464. [Google Scholar] [CrossRef]
- Murray, M.Y.; Birkland, T.P.; Howe, J.D.; Rowan, A.D.; Fidock, M.; Parks, W.C.; Gavrilovic, J. Macrophage migration and invasion is regulated by MMP10 expression. PLoS ONE 2013, 8, e63555. [Google Scholar] [CrossRef]
- Nighot, M.; Ganapathy, A.S.; Saha, K.; Suchanec, E.; Castillo, E.F.; Gregory, A.; Shapiro, S.; Ma, T.; Nighot, P. Matrix Metalloproteinase MMP-12 Promotes Macrophage Transmigration Across Intestinal Epithelial Tight Junctions and Increases Severity of Experimental Colitis. J. Crohns Colitis 2021, 15, 1751–1765. [Google Scholar] [CrossRef]
- Valdoz, J.C.; Johnson, B.C.; Jacobs, D.J.; Franks, N.A.; Dodson, E.L.; Sanders, C.; Cribbs, C.G.; van Ry, P.M. The ECM: To Scaffold, or Not to Scaffold, That Is the Question. Int. J. Mol. Sci. 2021, 22, 12690. [Google Scholar] [CrossRef] [PubMed]
- Kassianidou, E.; Probst, D.; Jäger, J.; Lee, S.; Roguet, A.-L.; Schwarz, U.S.; Kumar, S. Extracellular Matrix Geometry and Initial Adhesive Position Determine Stress Fiber Network Organization during Cell Spreading. Cell Rep. 2019, 27, 1897–1909.e4. [Google Scholar] [CrossRef] [PubMed]
- Estabridis, H.M.; Jana, A.; Nain, A.; Odde, D.J. Cell Migration in 1D and 2D Nanofiber Microenvironments. Ann. Biomed. Eng. 2018, 46, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Rübsamen, D.; Blees, J.S.; Schulz, K.; Döring, C.; Hansmann, M.-L.; Heide, H.; Weigert, A.; Schmid, T.; Brüne, B. IRES-dependent translation of egr2 is induced under inflammatory conditions. RNA 2012, 18, 1910–1920. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Klann, K.; Münch, C. Instrument Logic Increases Identifications during Multiplexed Translatome Measurements. Anal. Chem. 2020, 92, 8041–8045. [Google Scholar] [CrossRef]
- Schäfer, J.A.; Bozkurt, S.; Michaelis, J.B.; Klann, K.; Münch, C. Global mitochondrial protein import proteomics reveal distinct regulation by translation and translocation machinery. Mol. Cell 2022, 82, 435–446.e7. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuntschar, S.; Cardamone, G.; Klann, K.; Bauer, R.; Meyer, S.P.; Raue, R.; Rappl, P.; Münch, C.; Brüne, B.; Schmid, T. Mmp12 Is Translationally Regulated in Macrophages during the Course of Inflammation. Int. J. Mol. Sci. 2023, 24, 16981. https://doi.org/10.3390/ijms242316981
Kuntschar S, Cardamone G, Klann K, Bauer R, Meyer SP, Raue R, Rappl P, Münch C, Brüne B, Schmid T. Mmp12 Is Translationally Regulated in Macrophages during the Course of Inflammation. International Journal of Molecular Sciences. 2023; 24(23):16981. https://doi.org/10.3390/ijms242316981
Chicago/Turabian StyleKuntschar, Silvia, Giulia Cardamone, Kevin Klann, Rebekka Bauer, Sofie Patrizia Meyer, Rebecca Raue, Peter Rappl, Christian Münch, Bernhard Brüne, and Tobias Schmid. 2023. "Mmp12 Is Translationally Regulated in Macrophages during the Course of Inflammation" International Journal of Molecular Sciences 24, no. 23: 16981. https://doi.org/10.3390/ijms242316981