
NOD-Like Receptors: Guards of Cellular Homeostasis Perturbation during Infection


1
Institute of Immunology, Friedrich-Loeffler-Institut, 17493 Greifswald, Germany
2
Faculty of Mathematics and Natural Sciences, University of Greifswald, 17489 Greifswald, Germany
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(13), 6714; https://doi.org/10.3390/ijms22136714
Submission received: 14 May 2021
/
Revised: 14 June 2021
/
Accepted: 18 June 2021
/
Published: 23 June 2021
(This article belongs to the Special Issue The Diversity of Mammalian Nod-Like Receptor Functions)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The innate immune system relies on families of pattern recognition receptors (PRRs) that detect distinct conserved molecular motifs from microbes to initiate antimicrobial responses. Activation of PRRs triggers a series of signaling cascades, leading to the release of pro-inflammatory cytokines, chemokines and antimicrobials, thereby contributing to the early host defense against microbes and regulating adaptive immunity. Additionally, PRRs can detect perturbation of cellular homeostasis caused by pathogens and fine-tune the immune responses. Among PRRs, nucleotide binding oligomerization domain (NOD)-like receptors (NLRs) have attracted particular interest in the context of cellular stress-induced inflammation during infection. Recently, mechanistic insights into the monitoring of cellular homeostasis perturbation by NLRs have been provided. We summarize the current knowledge about the disruption of cellular homeostasis by pathogens and focus on NLRs as innate immune sensors for its detection. We highlight the mechanisms employed by various pathogens to elicit cytoskeleton disruption, organelle stress as well as protein translation block, point out exemplary NLRs that guard cellular homeostasis during infection and introduce the concept of stress-associated molecular patterns (SAMPs). We postulate that integration of information about microbial patterns, danger signals, and SAMPs enables the innate immune system with adequate plasticity and precision in elaborating responses to microbes of variable virulence.
Keywords:
innate immunity; NOD-like receptors; pathogens; cellular homeostasis; NOD1/2; NLRP3; NLRP11. Introduction
Nucleotide binding oligomerization domain (NOD)-like receptors (NLRs) are a group of evolutionarily conserved pattern recognition receptors (PRRs) critical for microbial recognition and host defense. To date, 22 NLRs have been identified in humans and 34 distinct ones in mice. They have a common molecular organization comprising a subclass-specific N-terminal effector domain, a central NOD domain, and C-terminal leucine-rich repeats (LRRs) that bind to pathogen-associated molecular patterns (PAMPs). Based on the distinct N-terminal effector domains including transactivator domain (AD), baculovirus inhibitor repeats (BIRs), caspase recruitment domain (CARD), or pyrin domain (PYD), NLRs are divided into four subgroups: NLRA, NLRB, NLRC, and NLRP [1,2]. The Major Histocompatibility Complex Class II Transactivator (CIITA), the only member of NLRA, induces expression of the major histocompatibility complex (MHC) class I and II by functioning as a transcriptional activator and a transcription factor, respectively [3,4]. The NLRB subgroup contains only one single member in humans, the NLR family apoptosis inhibitory protein (NAIP), and seven members (NAIP1-7) in mice. NAIP and NAIP1 sense needle proteins of the type III secretion system (T3SS) and activate the NLRC4 inflammasome, while NAIP5/6 and NAIP2 recognize bacterial flagellin and rod components, respectively [5,6,7,8]. The NLRC subgroup includes NOD1 and NOD2, which are well-characterized PRRs recognizing bacterial peptidoglycan. They sense γ-d-glutamyl-meso-diaminopimelic acid (iE-DAP) and muramyl dipeptide (MDP), respectively [9,10,11,12]. Ligand engagement leads to the release of auto-inhibitory conformation, subsequent oligomerization of NOD1/2 and recruitment of the receptor-interacting protein kinases 2 (RIP2) via CARD–CARD interactions, and finally activation of the nuclear factor kappa-light-chain-enhancer of activated B-cells (NF-κB) and mitogen-activated protein kinases (MAPKs), which drive expression of pro-inflammatory genes and antimicrobial responses [13,14]. The NLRP subgroup comprises 14 members in humans, NLRP3 being one of the best-investigated molecules in this subgroup and involved in inflammasome activation. NLRP3 is activated by diverse stimuli including PAMPs, damage or danger-associated molecular patterns (DAMPs) and cellular perturbations such as endoplasmic reticulum (ER) and mitochondrial stress. Upon activation, NLRP3 recruits the adaptor apoptosis-associated speck-like protein containing a CARD (ASC) and the pro-Caspase-1, triggering Caspase-1 activation and subsequent cleavage of interleukin 1 beta (IL1β) and eventually cell death [15,16]. The central NOD domain has an ATPase activity and is essential for the oligomerization of NLRs. Despite conservation of their global functions and signaling pathways, specific NLRs show differences between mice and humans. For instance, NOD1/2 trigger autophagy in both human and mouse cells, yet their downstream adaptor RIP2 is required for autophagy induction only in human cells [17,18]. The CARD only protein (COP) and the inhibitory CARD (INCA) are negative regulators of IL1β production and these are present solely in the human genome [19,20]. Moreover, lipopolysaccharide triggers an alternative inflammasome involving the Toll-like receptor 4 (TLR4)-RIP1-Caspase-8 signaling upstream of NLRP3, which is unique to human monocytes [21]. These examples illustrate the necessity for a careful consideration of potential similarities and differences in NLR biology across species. Specific examples are highlighted in the context of cell stress in this review.
Sensing of PAMPs by PRRs initiates immune responses irrespective of microbial pathogenicity. Detection of patterns of pathogenicity or homeostasis-altering molecular processes (HAMP) enables fine-tuning of immune responses and allows for differentiation of pathogens from nonpathogenic microbes [22,23]. During millions of years of co-evolution, pathogens have developed mechanisms targeting host homeostatic functions, and the resulting perturbations permit pathogens to infect, replicate and spread to permissive hosts. Accumulating evidence indicates that disruption of cellular homeostasis elicits NLR activation. Thus, we propose that in addition to PAMP sensing, NLRs also detect stress-associated molecular patterns (SAMPs), allowing for the discrimination of pathogens from harmless microbes. We extend the SAMP concept from intercellular stress signals (e.g., alerting neighboring cells of oxidative stress [24]) to intracellular cues that signify pathogenicity and activate innate immunity. Accordingly, cellular stress as a trigger and regulator of host antimicrobial defense will be discussed in the context of infection with various pathogenic microbes ranging from viruses to protozoans.
2. Pathogen-Induced Perturbation of Cellular Homeostasis
Although different pathogens employ a broad pool of virulence factors to perturb cellular homeostasis, the outcome of host–pathogen interactions converges toward conserved overarching cellular processes, notably cytoskeleton disruption, ER stress, mitochondrial dysfunction and protein translation inhibition.
2.1. Disruption of Cytoskeleton Dynamics
In eukaryotic cells, the cytoskeleton network comprises three components: actin filaments, tubulin microtubules and intermediate filaments [25]. It plays critical roles in diverse cellular processes such as endocytosis and phagocytosis, intracellular transport, cell migration and division [25]. The cytoskeleton network organization, especially its assembly and disassembly, is tightly and precisely regulated. However, pathogens have developed diverse strategies to manipulate the cytoskeleton dynamics at different stages of infection (Figure 1).
Under physiological conditions, in response to extracellular stimuli, the Rho family of GTPases including RHOA, RAC1, and CDC42 is turned on and subsequently induces activation of the nucleating factor actin related protein 2/3 (ARP2/3), which drives actin polymerization [26,27]. These pathways are exploited by pathogens for actin-mediated adhesion and invasion. For instance, extracellular bacteria such as enterohemorrhagic Escherichia coli (EHEC) and enteropathogenic E. coli (EPEC) translocate various effectors into cells via a type III secretion system (T3SS) to trigger actin polymerization and pedestal formation for bacterial adhesion. The translocated intimin receptor (Tir) is critical for the adhesion of EHEC and EPEC by activating the ARP2/3 complex [28,29,30]. The enteropathogenic E. coli effector protein F (EspF) binds to actin, profilin, ARP2 and to neural Wiskott-Aldrich syndrome protein (N-WASP), which subsequently induces actin polymerization [31]. EspG binds to tubulin, causing microtubule depolymerization and actin stress fiber formation [32]. In contrast, Clostridium difficile utilizes TcdA and TcdB toxins to inactivate the Ras homolog family member A (RHO), the Ras-related C3 botulinum toxin substrate 1 (RAC1) and the cell division control protein 42 homolog (CDC42) via glycosylation, thereby causing actin depolymerization and gut permeability [33,34]. Intracellular pathogens require access to subcellular compartments in phagocytic or non-phagocytic cells to meet their metabolic requirements for replication. Accordingly, various bacterial pathogens have developed strategies for rapid entry by inducing cytoskeleton reorganization. For example, Shigella flexneri secretes the invasion plasmid antigen C (IpaC) via T3SS to induce formation of filopodial extensions, activation of CDC42 and RAC1 for lamellipodial extensions, and of the avian sarcoma (Schmidt-Ruppin A-2) viral oncogene homolog (Src) kinase for actin polymerization, thus facilitating bacterial internalization [35,36,37]. Furthermore, Shigella utilizes the virulence factor A (VirA) to inhibit tubulin polymerization and destabilize microtubules for its efficient entry into epithelial cells [38,39]. Similar to Shigella, Salmonella typhimurium secretes several effectors via T3SS to activate CDC42 and RAC1 [40,41,42,43]. Salmonella outer protein E (SopE) and SopE2 trigger membrane ruffling and actin rearrangement by stimulating GDP/GTP exchange activity of CDC42 and RAC1 [40,41]. Salmonella invasion protein A (SipA) directly binds to actin and enhances actin polymerization [43]. Chlamydia trachomatis also secretes the effector called translocated actin recruiting phosphoprotein (TARP) to induce actin polymerization during entry into non-phagocytic cells [44]. Other intracellular bacteria including Listeria monocytogenes [45], Coxiella burnetii [46] and Yersinia pestis [47] do not utilize protein-mediated membrane ruffling. Instead, they invade host cells via an active zippering mechanism in which actin rearrangement is also involved [48].
Many viruses exploit the host cytoskeleton for their entry, replication, cellular transport and egress. Here, we only focus on mechanisms by which viruses actively induce cytoskeleton remodeling. For example, Herpes Simplex Virus-1 (HSV-1) induces RHOA activation and subsequently promotes its entry via an unusual phagocytosis-like uptake [49]. The Env protein of Human Immunodeficiency Virus (HIV) induces RAC1 activation and further promotes ARP2/3-dependent actin polymerization for HIV fusion with the cell membrane [50]. The Sendai virus increases the level of the actin-modifying protein Villin, leading to actin polymerization and viral entry [51]. Human Papillomavirus type 31 (HPV 31) activates tyrosine and phosphoinositide 3-kinases (PI3K) to promote cytoskeletal rearrangement, which allows viral entry via filopodia transport [52].
For obligate intracellular parasites, plasma membrane and cytoskeleton form barriers against invasion, hence some parasites actively induce actin reorganization to facilitate their entry and release. Toxoplasma gondii and Plasmodium berghei trigger the formation of ring-shaped actin structures at the parasite–cell junction for host cell invasion [53]. T. gondii secretes the actin-binding protein toxofilin to regulate actin filament disassembly and turnover [54], whereas P. falciparum employs several proteases such as serine protease gp76 [55], chymotrypsin-like protease [56], plasmepsin II [57] and possibly the cysteine protease [58] to induce actin rearrangement by cleavage of various actin-associated factors [56,57,58].
After cell invasion, several pathogens hijack the cytoskeleton network to subvert intracellular killing mechanisms. Shigella induces the formation of a unique actin coat-like structure that inhibits bacterial killing by blocking fusion of the phagosomes with late endosomes/lysosomes [59,60]. This actin cocoon is triggered by the T3SS effector IcsB (Intra-inter-cellular spread B) by recruiting the actin nucleation machinery [60]. Massive cytoskeleton perturbation also occurs during intracellular replication of C. trachomatis. Chlamydia inclusions, which contain replicating bacteria, are enclosed in compact F-actin and intermediate filaments that cooperatively maintain integrity and stability of the inclusions [61]. C. burnetii replicates in large parasitophorous vacuoles that require F-actin association in a process depending on RHOA and CDC42 [62].
Several pathogens take advantage of actin-based motility for cell-to-cell spread. L. monocytogenes achieves such motility via the bacterial factor actin assembly-inducing protein (ActA) [63]. ActA mimics the ARP2/3 nucleation-promoting factor N-WASP. It binds to actin monomers and activates ARP2/3, thus promoting actin nucleation [64,65]. IcsA from Shigella recruits N-WASP and ARP2/3 on its surface to induce actin polymerization [66,67,68]. Rickettsia parkeri utilizes two actin-polymerizing proteins, RickA and surface cell antigen 2 (Sca2), for early and late motility, respectively [69]. Various Burkholderia species also show distinct actin-based motility mediated by Burkholderia intracellular motility A (BimA) orthologs by mimicking different host actin-polymerizing proteins [70]. The actin tails formed at the bacterial surface generate mechanical forces that propel the bacteria into adjacent cells via membrane protrusion [71]. Several viruses also manipulate the actin cytoskeleton to induce rapid cell-to-cell spread without free virion release. The Vaccinia virus employs the viral protein A36 [72,73] and the Baculovirus uses the P78/83 capsid protein [74] to promote ARP2/3-dependent actin polymerization. Actin tails are also found to be associated with Ebola virus nucleocapsids, contributing to its budding [75]. Pseudorabies virus (PRV) and HSV-1 induce cytoskeletal rearrangements and cell extensions to facilitate viral spread by US3 kinase [76,77]. The P protein of Human Metapneumovirus (HMPV) promotes CDC42, RAC1 and RHOA dependent formation of intercellular actin extensions, thus contributing to direct cell-to-cell spread [78].
In sum, bacterial, viral and protozoal pathogens employ unrelated virulence factors that target actin, tubulin and intermediate filaments to induce host cytoskeleton remodeling for their invasion, intracellular survival and exit from host cells. The dysregulated cytoskeleton dynamics results in the perturbation of homeostatic functions and signifies pathogenicity during infection.
2.2. ER Stress
The ER maintains cellular homeostasis by regulating protein folding and processing, lipid synthesis and calcium storage and release. Accumulation of misfolded proteins in the ER lumen leads to its functional impairment and ER stress [79,80]. To counteract the detrimental effects of the ER stress, eukaryotic cells initiate the unfolded protein response (UPR) pathways. The ER stress is sensed by three ER transmembrane receptors: protein kinase R-like ER kinase (PERK), inositol-requiring enzyme 1α (IRE1α) and activating transcription factor 6 (ATF6). Under homeostatic conditions, these sensors are inactive due to interactions with the ER luminal heat shock protein 70 (HSP70)-type chaperone binding immunoglobulin protein (BiP). Upon ER stress, BiP dissociates from these sensors and binds to misfolded proteins in the ER, thereby releasing the sensors to initiate the three branches of UPR [79,80].
Diverse pathogens disrupt ER homeostasis and cause ER stress, thus facilitating their survival and replication and controlling host cell death (Figure 2). Viruses exploit the ER for their protein synthesis and processing. Hence, a large number of viruses can induce ER stress to promote their replication, twist host cell death and facilitate their dissemination [81]. For instance, the African Swine Fever Virus (ASFV) utilizes ER as replication sites. It activates ATF6-dependent UPR to promote viral replication [82,83]. The Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) induces activation of PERK via its spike [84] and 3a protein [85] as well as the activation of ATF6 via the 8ab protein [86]. Dengue Virus (DENV) triggers IRE1α, PERK and ATF6-mediated UPR to promote virion assembly and alleviate virus-induced apoptosis [87,88,89,90].
Various bacteria and parasites also trigger ER stress, contributing to the pathogenesis of infection. The 6 kDa early secretory antigenic target (ESAT-6), a secreted virulence factor from Mycobacterium tuberculosis (Mtb), activates IRE1α and PERK mediated UPR, resulting in Ca2+ release, reactive oxygen species (ROS) production, and subsequent apoptosis [91]. Heparin-binding hemagglutinin antigen (HBHA) from Mtb also induces ER stress through cytosolic Ca2+ and ROS generation, leading to apoptosis [92]. Shiga toxin 1 from S. dysenteriae, the cholesterol-dependent cytolysin Listeriolysin O (LLO) from L. monocytogenes as well as EHEC and C. trachomatis activate all ER stress sensors IRE1α, PERK, and ATF6 [93,94,95,96]. B. abortus resides in ER-derived vacuoles and VceC, an effector of T4SS, directly interacts with the ER chaperon BiP and activates the IRE1α- X-box binding protein 1 (XBP1) pathway [97]. The cyclic dinucleotide c-di-GMP from B. abortus also triggers stimulator of interferon genes (STING)-dependent ER stress responses, which facilitate bacterial replication in vivo [98]. Streptolysins O and S from group A Streptococcus (GAS) elicit ER stress, and promote biofilm formation and dissemination within soft tissues in vivo [99]. C. burnetii stimulates IRE1α signaling and inhibits ER stress-induced apoptosis via the T4SS effector CaeB (C. burnetii anti-apoptotic effector B), likely contributing to pathogenicity in vivo [100]. P. berghei induces IRE1α-mediated UPR, promoting liver stage infection [101], while T. gondii activates the same ER stress sensor to enhance its dissemination in vivo [102]. Altogether, viral, bacterial or parasitic pathogens often induce ER stress to promote their replication and dissemination. Their abilities to induce ER stress distinguish them from non-pathogenic microbes.
2.3. Mitochondrial Dysfunction
Mitochondria are central organelles for many homeostatic processes including oxidative phosphorylation and ATP production, fatty acid oxidation and cell death. They are also crucial for innate immunity and inflammatory responses [103,104]. RNA sensors (e.g., retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated protein 5 (MDA5)) converge their signals toward mitochondrial antiviral-signaling protein (MAVS), which triggers TANK-binding kinase 1 (TBK1)/Interferon regulatory factor 3 (IRF3)-dependent type I interferon (IFN-I) signaling [105,106,107,108]. Mitochondria-derived ROS also facilitate bacterial killing [109,110]. Moreover, metabolic reprogramming of immune cells in mitochondria governs immune responses and inflammation [111,112,113,114]. Therefore, it is not surprising that many pathogens have developed strategies to provoke mitochondrial dysfunction (Figure 3) and thus escape the host defense.
Mitochondria are dynamic organelles that constantly undergo fission and fusion and their dynamics are important for processes such as energy production, mitochondrial quality control and cell death. Manipulation of mitochondrial dynamics, either fragmentation or elongation, has been reported as pathogenicity mechanism for bacterial and viral pathogens (Figure 3A). L. monocytogenes elicits mitochondrial fragmentation via multiple mechanisms. The pore-forming toxin LLO causes loss of mitochondrial membrane potential and reduces ATP generation [115], increases expression of MIC10, a critical component of the mitochondrial contact site and cristae organizing system (MICOS) complex [116], and induces mitochondrial fission independent of the dynamin related protein 1 (DRP1) [117]. Altering dynamics of mitochondrial fusion during infection affects survival of Listeria, suggesting that it manipulates mitochondria dynamics to establish an infection [115]. B. abortus induces a drastic mitochondrial fragmentation independent of DRP1 [118], whereas H. pylori utilizes the vacuolating cytotoxin A (VacA) to elicit mitochondrial fragmentation via a mechanism dependent on DRP1 [119]. Similarly, S. flexneri and L. pneumophila induce DRP1-dependent mitochondrial fragmentation for their intracellular replication [120,121], whereas Influenza A Virus (IAV) employs the viral protein PB1-F2 to cause such fragmentation. Moreover, PB1-F2 is imported to mitochondria via the translocase of the outer membrane 4 (TOM4) and inhibits RIG-I/MVAS-mediated IFN-I response [122]. Other viral pathogens rather stimulate mitochondrial fusion. For instance, the open reading frame-9b (ORF-9b) from SARS-CoV triggers ubiquitination and proteasomal degradation of DRP1, resulting in mitochondrial elongation, degradation of MAVS, TNF receptor-associated factor (TRAF3) and TRAF6 and inhibition of IFN-I [123]. The glycoprotein 120 (gp120) of HIV induces mitochondrial fusion by reducing total and active DRP1 levels [124].
Various pathogens are also reported to modulate the host metabolism to ensure a replication niche (Figure 3B). Mtb induces upregulation of key glycolytic enzymes while lowering expression of enzymes involved in the tricarboxylic acid (TCA) cycle and oxidative phosphorylation in murine lungs, suggesting a metabolic shift from oxidative phosphorylation to aerobic glycolysis in tuberculosis [125]. Mtb also increases glycolysis in human and mouse macrophages in vitro, in turn contributing to bacterial control via IL1β release [126]. This metabolic shift toward glycolysis is initiated by the toll-like receptor 2 (TLR2) activation and mediated by the protein kinase B/mechanistic target of the rapamycin (AKT/mTOR) pathway [127]. Mtb augments expression of microRNA-21 (miR-21) in macrophages, which in turn dampens glycolysis by targeting phospho-fructo-kinase, muscle isoform (PFK-m). IFNγ enhances PFK-m expression, thereby contributing to improved control of Mtb [128]. Opposingly, Mtb promotes a quiescent energy phenotype with reduced glycolysis and TCA cycle in human monocyte-derived macrophages, while attenuated M. bovis BCG or dead Mtb induce glycolysis [129]. These apparently contradictory findings may be attributed to host-, cell type-, or infection stage-specific metabolic remodeling during Mtb infection. Other intracellular bacteria (e.g., L. pneumophila and B. abortus) trigger a metabolic switch toward glycolysis to support bacterial survival [121,130]. During S. flexneri infection, host cells convert glucose into acetate instead of lactate, and the bacteria capture the downstream metabolite pyruvate for their growth [131]. Similarly, the Apicomplexa parasite Theileria induces aerobic glycolysis in bovine leukocytes [132,133] by stabilizing host pyruvate kinase isoform M2 (PKM2) [134]. To utilize host glucose as a carbon source, T. gondii also promotes glycolysis in murine dendritic cells [135] and in fibroblasts [136,137] by increasing the expression of enzymes involved in glycolysis [135,136,137]. T. cruzi enhances host glucose uptake to fuel its metabolism, yet without affecting glycolysis [138]. Instead, the protozoa P. falciparum hampers glycolysis in red blood cells due to oxidative stress [139]. Viruses completely rely on host metabolism to fuel their replication, however, some of them are able to actively subvert host metabolism for their benefit. The infection with the Human Cytomegalovirus (HCMV) increases the glycolytic flux, while HSV-1 induces the TCA cycle for its replication [140]. The Hepatitis C Virus (HCV) infection increases glycolysis and activates the pentose phosphate pathway as well as lipid synthesis for virus replication at the early stage, and fatty acid oxidation at later stages [141].
Overall, these findings strengthen the concept that mitochondrial dysfunction represents an evasion mechanism that facilitates efficient escape from host defense during viral, bacterial and protozoal infection.
2.4. Blockade of Protein Translation
Viral replication completely depends on the host’s translation machinery. To facilitate translation of viral proteins and restrict production of host antiviral proteins, viruses have developed multiple strategies to globally block host translation (Figure 3C). Translation of host mRNA occurs in a cap-dependent manner and the eukaryotic initiation factor 4F (eIF4F) complex consisting of the cap-binding protein eIF4E, the scaffold eIF4G, and the RNA helicase eIF4A is critical for efficient targeting of ribosomes to the m7G cap [142,143]. Many viruses directly target the eIF4F complex given the importance of the complex in host mRNA translation [144]. Poliovirus [145], coxsackievirus and rhinovirus [146], HIV-1 [147], HIV-2, moloney murine leukemia virus, mouse mammary tumor virus and simian immunodeficiency virus [148], feline calicivirus [149] and foot-and-mouth disease virus [150] encode various proteases to directly degrade eIF4G. The rotavirus non-structural protein 3 (NSP3), a homolog of the poly(A) binding protein (PABP), interacts with eIF4G and disrupts interaction between eIF4G and PABP, thus shutting off host protein synthesis while keeping viral protein translation unaltered [151]. Enteroviruses induce expression of miR-141 that targets eIF4E mRNA for its degradation [152]. NSP1 from SARS-CoV-2 binds to the 40S ribosomal subunit, leading to the blockade of mRNA entry and disruption of host translation [153].
Although bacteria do not depend on the host translation machinery, some species also develop strategies to impair host translation and inhibit the immune defense (Figure 3C). Exotoxins (e.g., diphtheria toxin from C. diphtheriae, shiga toxin from S. dysenteriae and exotoxin A from P. aeruginosa) block protein translation by inactivating the host elongation factor 2 (EF2) via ADP-ribosylation [154]. C. trachomatis substantially inhibits host protein synthesis, possibly by inducing ER or mitochondrial stress [155]. L. pneumophila uses several effectors to interfere with host protein translation. Legionella glucosyltransferase 1 (Lgt1) inactivates the elongation factor 1-alpha (EF1A) [156], the substrate I of Icm/Dot transporter (SidI) binds to and disables EF1A and EF1Bγ [157], and Legionella kinase 4 (LegK4) phosphorylates all members of the HSP70 family (HSP70, HSP72, and BiP) and compromises folding of nascent polypeptides [158,159]. Collectively, although host translation is primarily targeted by viruses, selected bacterial pathogens employ multiple toxins or effectors to inhibit protein translation.
3. NLRs Activation by Perturbation of the Cellular Homeostasis
Several NLR members can be activated by PAMPs, DAMPs and also by pathogen-induced perturbation of cellular homeostasis, possibly via SAMP generation. Members of two NLR subclasses, NLRC and NLRP, are major receptors involved in inflammation induced by disrupted cellular homeostasis during pathogen invasion. Involvement of NOD1 and NOD2 as well as of NLRP1 and NLRP3 in sensing cellular homeostasis dysfunction has been recently demonstrated [160,161,162,163]. These NLR members are endowed with remarkable multitasking abilities of directly sensing microbial moieties, detecting danger cues and monitoring cellular stress during infection.
3.1. NOD1 and NOD2 Detect the Disruption of Cytoskeleton Dynamics
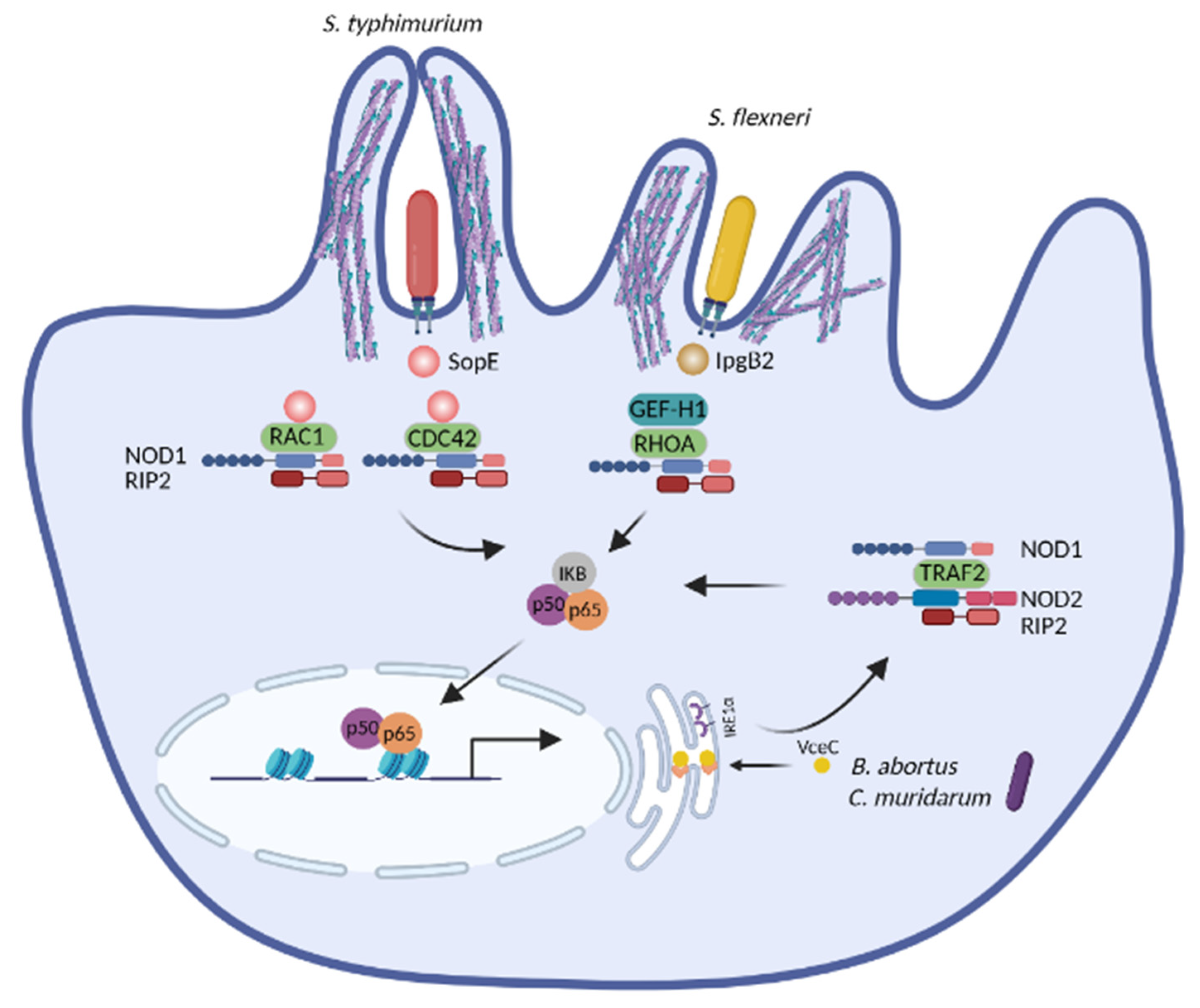
Several studies have unveiled that NOD1 and NOD2 are activated upon cytoskeleton disruption [164,165,166,167]. Experiments with chemical inhibitors have first indicated that actin-depolymerizing agents (e.g., cytochalasin D and latrunculin B) and actin-polymerizing compounds (e.g., jasplakinolide) induce NF-κB activation and IL8 production in a process depending on NOD2 [164,165,168]. RAC1 interacts with NOD2 and expression of a RAC1 dominant negative mutant leads to loss of membrane ruffles and dissociation of NOD2 from RAC1, further enhancing NOD2-mediated NF-κB activation [164]. Consistently, expression of the dominant negative RAC1 mutant triggers NOD2/RIP2-dependent NF-κB activation [169]. Many pathogens manipulate RHO GTPases to induce cytoskeleton rearrangement and host invasion [36,38,40,41,42] and these processes promote NOD1 and/or NOD2 mediated immune activation (Figure 4). The T3SS effector SipA from Salmonella requires NOD1 and NOD2 as well as their downstream adaptor RIP2 to trigger NF-κB activation. The actin-binding domain of SipA is dispensable for NF-κB activation, suggesting that its ability to cause actin rearrangement is obsolete for the NF-κB effects [166]. SipA also induces redistribution of LAMP1-positive compartments toward the microtubule-organizing center (MTOC) [170]. Thus, SipA-induced NOD1 and NOD2 activation is likely connected with its ability to manipulate the microtubule network. Another T3SS effector, SopE from Salmonella, a nucleotide-exchange factor, causes membrane ruffling by activating RHO GTPases [40]. It further promotes the interaction of NOD1 with RAC1 and CDC42, which in turn triggers NOD1/RIP2-mediated NF-κB activation. In this case, NOD1 likely senses bacteria-induced aberrant activation of RHO GTPases [167]. Another cytoskeleton-related molecule activating NOD1 is the guanine nucleotide exchange factor H1 (GEF-H1). GEF-H1 stimulates the formation of actin stress fibers by activating RHOA upon its dissociation from microtubules [171] and is required for NOD1 canonical ligand-induced NF-κB activation, however, independent of RHOA [172]. GEF-H1 also contributes to NOD2-induced NF-κB activation and cytokine production by mediating the phosphorylation of RIP2 [173]. In the context of S. flexneri infection, IpgB2, a T3SS effector, binds to the mammalian homolog of Diaphanous (mDia) and directly induces actin polymerization by functioning as an analog of active RHOA [35]. In line with its role in actin polymerization, IpgB2 localizes to actin-associated cellular junctions when expressed in cells. Ectopic expression of the invasion plasmid gene B2 (IpgB2) or outer Shigella protein B (OspB), another T3SS effector without a known role in cytoskeleton remodeling, induces GEF-H1 and NOD1-mediated NF-κB activation, yet is independent of the classical NOD1/2 downstream signaling component RIP2 [172]. Other molecules interfering with NOD1 activation are Cofilin and Cofilin phosphatase slingshot homolog 1 (SSH1), which control the disassembly of actin filaments. Both are essential for maintaining actin cytoskeleton dynamics and inducing NF-κB activation upon stimulation with Shigella and NOD1 agonists [174]. As RHOA negatively regulates Cofilin, it seems unlikely that NOD1 activation is connected with excessive RHO activation under these conditions.
Altogether, pathogens exploit various mechanisms to hijack the cytoskeleton dynamics. During infection, NOD1 and/or NOD2 represent an intracellular surveillance system inducing NF-κB activation by detecting cytoskeleton disruption, instead of monitoring RHO GTPases. The detailed mechanisms by which NOD1 and NOD2 monitor cytoskeleton dynamics and induce NF-κB activation need further elucidation.
3.2. NOD1 and NOD2 Sense the ER Stress during Infection
Chemical agents that cause ER stress such as tunicamycin, brefeldin A, 2-deoxyglucose and thapsigargin (TG) trigger an NF-κB-dependent inflammation. However, the ER stress induced UPR is uncoupled from NF-κB activation, suggesting that ER stress activates two distinct signaling pathways: the classical UPR and the NF-κB activation [175]. Further investigations have linked ER stress-induced NF-κB activation with the intracellular PRRs NOD1/2 [160]. IL6 production upon TG stimulation is impaired by NOD1/2 knockout (KO), demonstrating NOD1/2 activation by ER stress. The ER stress inhibitor tauroursodeoxycholic acid (TUDCA) and the IRE1α inhibitor KIRA6 (IRE1α kinase inhibiting RNase attenuator 6) block TG-induced IL6 production in vitro and in vivo, while both inhibitors have no impact on IL6 production stimulated by MDP, the canonical bacterial agonist of NOD2. Thus, the ER stress and bacterial PAMPs employ different signaling pathways to activate NOD1/2 [160]. In the context of infection, the B. abortus T4SS effector virB-coregulated effector C (VceC) induces ER stress through interaction with the chaperon BiP [97] and triggers NOD1/2- and RIP2-dependent IL6 production. Infection of NOD1/2 KO mice with wildtype of B. abortus or infection of wildtype mice with the VceC null mutant yields decreased IL6 production, milder pathology and increased survival of mice. Therefore, the ER stress elicited by B. abortus infection activates NOD1/2-dependent inflammation, hence contributing to the pathogenesis of brucellosis [160]. C. muridarum infection also induces ER stress and NOD1/2-RIP2-mediated IL6 production [160], however, inhibition of ER stress or depletion of NOD1/2 or RIP2 leads to increased bacterial burdens in vivo. Thus, the ER stress and subsequent NOD1/2-dependent inflammation caused by Chlamydia opposingly contributes to infection clearance [96]. Furthermore, the ER stress induced by TG significantly increases NF-κB activation and the expression of IL6 and IL23 in response to NOD1 stimulation upon S. enterica infection in vitro. However, the impact of the ER stress during S. enterica infection in vivo remains to be elucidated [176]. Together, NOD1 and NOD2 activate inflammatory responses by detecting the ER stress caused by various pathogens (Figure 4), yet the outcome of ER stress-induced NOD1/2 activation is disease-dependent. Given that many pathogens induce ER stress [81] and that peptidoglycan-free viruses and parasites activate NOD1 and/or NOD2-dependent inflammation [177], it is reasonable to speculate that NOD1 and/or NOD2 are activated by sensing the ER stress and possibly SAMPs during these infections. Additionally, aberrant ER stress is associated with many noncommunicable diseases such as neurodegenerative disorders, atherosclerosis, type 2 diabetes, and cancers [178]. Hence, it is worthwhile to further investigate whether the ER stress-induced inflammation in patients suffering from these diseases confers susceptibility to specific infections.
The molecular events that integrate ER stress with NOD1/2 signaling and NF-κB activation are a focus of current research. Upon ER stress, IRE1α binds to TRAF2, induces its oligomerization and the activation of NF-κB and c-Jun N-terminal kinases (JNK) [160,179]. Although NOD1 and NOD2 contain motifs predicted to bind TRAF2, evidence for a direct interaction of TRAF2 with NOD1/2 is missing. Moreover, whether kinase activity of IRE1α or other factors alter the putative interaction of TRAF2 and NOD1/2 awaits to be elucidated. A study demonstrates that ER stress triggered by TG results in Ca2+ efflux and subsequent Ca2+-dependent NOD1/2-mediated inflammation. Peptidoglycan contaminants in the serum could be internalized via endocytosis upon ER stress and subsequently activate NOD1 and NOD2 [180]. However, whether such trace amounts of peptidoglycan, or possibly other ligands, in the serum activate NOD1/2 requires further investigations. Recently, we have uncovered that generation of the endogenous metabolite sphingosine-1-phosphate (S1P) is remarkably increased upon various types of stress including ER stress and cytoskeleton disruption elicited by chemicals. However, other metabolites in the sphingolipid pathway (i.e., Ceramide and Sphinganine) are not affected by the same stimulations. S1P, but not Ceramide, directly binds to NOD1 and NOD2, and S1P delivery into cells leads to NOD1 or NOD2 mediated NF-κB activation. Hence, we propose that NOD1/2 detect perturbation of cellular homeostasis through sensing of the cytosolic metabolite S1P, which represents a SAMP [181]. Whether the S1P-NOD1/2 axis is activated during infection with peptidoglycan-free pathogens needs to be elucidated.
3.3. Activation of the NLRP3 Inflammasome by the ER Stress
Multiple studies have revealed that the ER stress triggered by chemical agents induces NLRP3 inflammasome activation, however, NLRP3 activation elicited by pathogen-induced ER stress has rarely been reported. Chemically induced ER stress (i.e., by tunicamycin or brefeldin A) causes activation of the NLRP3 inflammasome. This effect is uncoupled from the stress sensors IRE1α, PERK and ATF6α, but is dependent on ROS and potassium efflux that are mediated by the voltage-dependent anion-selective channel 1 (VDAC1), which transports metabolites and ions into mitochondria. Thus, ER stress-induced inflammasome activation and UPR are divergent processes under these circumstances [182]. Mechanistically, the ER stress induces the expression of thioredoxin-interacting protein (TXNIP) via PERK and IRE1α-mediated signaling pathways. TXNIP induces IL1β mRNA transcription and further activates the NLRP3 inflammasome for IL1β release [183]. Moreover, the ER stress also increases TXNIP mRNA by reducing the levels of the TXNIP destabilizing micro-RNA miR-17 via IRE1α [184] and by ROS formation [185], further leading to NLRP3 inflammasome activation. Thus, the IRE1α-TXNIP axis mediates activation of the NLRP3 inflammasome-subsequent ER stress, and might serve as a therapeutic target for metabolic disorders [186,187]. In the context of infection, B. abortus induces ER stress and subsequently NLRP3 inflammasome activation. In this circumstance, TXNIP expression is elevated and it translocates to mitochondria, resulting in increased mitochondrial ROS and mitochondrial damage, which result in NLRP3 inflammasome activation [188] (Figure 5A). Collectively, the chemical- or pathogen-induced ER stress activates the NLRP3 inflammasome via TXNIP, a process involving mitochondrial alterations. Whether other pathogens cause ER stress and this type of cell stress engages similar molecular pathways for IL1β production remains to be investigated.
3.4. NLRP3 Inflammasome Induced by Mitochondrial Dysfunction
Mitochondrial dysfunction leads to activation of the NLRP3 inflammasome [189,190] (Figure 5B). Inhibition of complex I, one of the key enzymes of the respiratory chain, results in robust production of mitochondrial ROS causing NLRP3 inflammasome activation [163]. VDAC1/2, which is critical for mitochondrial ROS generation, is required for the inflammasome induction by inhibiting complex I [163]. Consistently, stimulation with the TLR7 ligands imiquimod and CL097 results in ROS production and activation of the NLRP3 inflammasome [191]. Mitophagy is essential for maintaining mitochondrial homeostasis [192]. Defects in autophagy induce accumulation of damaged mitochondria and elevated NLRP3 inflammasome activation that is dependent on both mitochondrial ROS and release of mitochondrial DNA (mtDNA) in the cytosol [190,193,194]. Mitochondrial dysfunction also induces release of oxidized mtDNA, which directly binds to and activates the NLRP3 inflammasome [195]. The replication of mtDNA is dependent on the cytidine/uridine monophosphate kinase 2 (CMPK2), which further contributes to the production of oxidized mtDNA and subsequent NLRP3 activation [196]. In the context of infection, the T3SS effector Salmonella invasion protein B (SipB) from S. typhimurium, localizes to mitochondria causing their swelling and depolarization, which subsequently induces oxidized mtDNA-mediated NLRP3 inflammasome [195,197]. Some antibiotics also disturb mitochondrial functions due to the resemblance between mitochondria and bacteria. Linezolid, an oxazolidinone antibiotic, induces NLRP3 inflammasome activation by promoting mitochondrial dysfunction. The mitochondrial lipid cardiolipin recruits NLRP3 to the mitochondria by direct binding and stimulates NLRP3 activation [198]. Collectively, mitochondrial dysfunction triggers NLRP3 inflammasome activation via mitochondrial ROS, mtDNA, oxidized mtDNA or cardiolipin. Whether such pathways generally participate in pathogen-induced NLRP3 inflammasome activation requires further elucidation.
3.5. NLRP1 Inflammasome and Cellular Homeostasis
The mouse NLRP1B (mNLRP1B) was first identified as a critical component conferring susceptibility to the anthrax lethal factor (LF). LF-induced cell death is mediated by Caspase-1, suggesting that mNLRP1B mediates pyroptosis by LF [199]. The N-terminal proteolysis is crucial for LF-induced mNLRP1B inflammasome activation [200]. Similarly, the 3C protease of human rhinovirus directly cleaves and activates human NLRP1 (hNLRP1) inflammasome [162]. After cleavage by LF, mNLRP1B generates a new N-terminus recognized by UBR2, leading to the ubiquitination of mNLRP1B and degradation by the N-end rule pathway, which are critical for mNLRP1B inflammasome activation [161,201]. The Shigella effector invasion plasmid antigen H7.8 (IpaH7.8), an E3 ubiquitin ligase, also activates mNLRP1B inflammasome by directly ubiquitinating mNLRP1B for degradation [161]. In the case of hNLRP1, the glycine-specific N-degron machinery cullinZER1/ZYG11B mediates recognition and degradation of hNLRP1 subsequent cleavage by the 3C protease of human rhinovirus [162]. Therefore, functional degradation of NLRP1 allows the host immune system to distinguish pathogens by monitoring the downstream cellular damage [202]. In addition to pathogen-induced activation, Val-boroPro (VbP), an inhibitor of dipeptidyl peptidases 8 and 9 (DPP8/9), also triggers both mNLRP1B and hNLRP1 inflammasome activation [203]. Both the peptidase activity of DPP9 and its binding to hNLRP1 are required for hNLRP1 activation by VbP [204]. Therefore, it has been proposed that hNLRP1 may indirectly sense the perturbation of cellular homeostasis induced by DPP8/9 cleavage [205]. In line with this, 2-deoxyglucose (2DG), a glycolysis inhibitor, and sodium azide, an inhibitor of the mitochondrial electron transport chain, activate the mNLRP1B inflammasome, suggesting a link between ATP production and mNLRP1B activation. Indeed, ATP depletion by glucose starvation or hypoxia induces the activation of the mNLRP1B inflammasome [206]. Altogether, NLRP1 may sense the disruption of cellular homeostasis induced by DPP8/9 cleavage, cellular ATP depletion, or infections (Figure 5C). Elucidation of the substrate of DPP8/9 will shed light on the mechanism of NLRP1 activation by disruptions of cellular homeostasis.
4. Conclusions and Perspectives
Solely detecting PAMPs or DAMPs appears insufficient for accurate identification of pathogens by the immune system. First, PAMPs are present in commensal, pathobionts, and pathogenic microorganisms. Second, DAMPs can be produced upon pathogen-induced damage or during sterile tissue damage. DAMPs such as ATP, F-actin and DNA binding proteins (e.g., High-mobility group box 1 (HMGB1)) are host molecules released from damaged cells [207]. Hence, the immune system requires additional mechanisms to specifically detect the different types of damage and accordingly fine-tune the antimicrobial responses. Pathogens subvert host defense mechanisms by manipulating various cellular pathways, which in turn often leads to perturbation of cellular homeostasis. In plants, NLRs recognize directly or indirectly pathogen effector proteins or abiotic stress, resulting in effector-triggered immunity [208]. In line with this, detection of perturbations of cellular homeostasis by NLRs may also represent an ancient mechanism of sensing infection in animals. Detection of perturbations of cellular homeostasis enables the immune system to distinguish pathogens from nonpathogenic microbes and adjust the magnitude of immune response. Depending on the extent and types of stress, the innate immune system could activate different pathways, for instance, NOD1/2-mediated NF-κB activation and NLRPs-mediated pyroptosis. Thus, according to the severity of the damage, the innate immune system could adapt and apply appropriate host defense stratagems.
It remains elusive how NLRs detect diverse and broad types of cellular perturbation. Similarly, it is unclear how NLRs adapt to different types of cellular dysfunction. It has been postulated that NLRs could maintain inactive states by means of host decoy or guard proteins and that modifications of these proteins induced by pathogens trigger the activation of NLRs [209]. In the same direction, it is reasonable to hypothesize that cellular homeostasis dysfunction could modify host proteins or their activities and thereby activate NLRs. Indeed, generation of S1P is increased upon perturbation of cellular homeostasis and subsequently S1P triggers NOD1/2-medidated activation of innate immunity [181]. Considering that cellular homeostasis disruption induced by pathogens converges toward conserved pathways and molecules, these altered proteins/metabolites are designated as SAMPs. Accordingly, NLRs could detect the perturbation of cellular homeostasis by monitoring SAMPs such as S1P. The identities of SAMPs activating other NLRs upon distinct types of cellular stress, how SAMPs are generated and the distinct mechanisms employed by NLRs to differentially detect SAMPs, PAMPs or DAMPs remain to be elucidated. Given that many SNPs in NLRs are associated with autoimmune diseases, the SAMPs–NLRs axis likely represents an evolutionary conserved mechanism involved in infection, sterile inflammation and autoimmune disorders. Comprehensive understanding of the mechanisms of NLRs activation will help to harness the power of NLRs to fight infections by boosting inflammatory responses or reducing excessive inflammation in NLRs-associated autoinflammatory disorders.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank Robert Golinski for editorial assistance.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kanneganti, T.D.; Lamkanfi, M.; Nunez, G. Intracellular NOD-like receptors in host defense and disease. Immunity 2007, 27, 549–559. [Google Scholar] [CrossRef]
- Motta, V.; Soares, F.; Sun, T.; Philpott, D.J. NOD-like receptors: Versatile cytosolic sentinels. Physiol. Rev. 2015, 95, 149–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, B.K.; Chin, K.C.; Olsen, J.C.; Skinner, C.A.; Dey, A.; Ozato, K.; Ting, J.P. Induction of MHC class I expression by the MHC class II transactivator CIITA. Immunity 1997, 6, 591–600. [Google Scholar] [CrossRef] [Green Version]
- Masternak, K.; Muhlethaler-Mottet, A.; Villard, J.; Zufferey, M.; Steimle, V.; Reith, W. CIITA is a transcriptional coactivator that is recruited to MHC class II promoters by multiple synergistic interactions with an enhanceosome complex. Genes Dev. 2000, 14, 1156–1166. [Google Scholar] [PubMed]
- Yang, J.; Zhao, Y.; Shi, J.; Shao, F. Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc. Natl. Acad. Sci. USA 2013, 110, 14408–14413. [Google Scholar] [CrossRef] [Green Version]
- Rayamajhi, M.; Zak, D.E.; Chavarria-Smith, J.; Vance, R.E.; Miao, E.A. Cutting edge: Mouse NAIP1 detects the type III secretion system needle protein. J. Immunol. 2013, 191, 3986–3989. [Google Scholar] [CrossRef]
- Lightfield, K.L.; Persson, J.; Brubaker, S.W.; Witte, C.E.; von Moltke, J.; Dunipace, E.A.; Henry, T.; Sun, Y.H.; Cado, D.; Dietrich, W.F.; et al. Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat. Immunol. 2008, 9, 1171–1178. [Google Scholar] [CrossRef]
- Rauch, I.; Tenthorey, J.L.; Nichols, R.D.; Al Moussawi, K.; Kang, J.J.; Kang, C.; Kazmierczak, B.I.; Vance, R.E. NAIP proteins are required for cytosolic detection of specific bacterial ligands in vivo. J. Exp. Med. 2016, 213, 657–665. [Google Scholar] [CrossRef]
- Girardin, S.E.; Boneca, I.G.; Carneiro, L.A.; Antignac, A.; Jehanno, M.; Viala, J.; Tedin, K.; Taha, M.K.; Labigne, A.; Zahringer, U.; et al. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science 2003, 300, 1584–1587. [Google Scholar] [CrossRef] [Green Version]
- Girardin, S.E.; Boneca, I.G.; Viala, J.; Chamaillard, M.; Labigne, A.; Thomas, G.; Philpott, D.J.; Sansonetti, P.J. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J. Biol. Chem. 2003, 278, 8869–8872. [Google Scholar] [CrossRef] [Green Version]
- Girardin, S.E.; Travassos, L.H.; Herve, M.; Blanot, D.; Boneca, I.G.; Philpott, D.J.; Sansonetti, P.J.; Mengin-Lecreulx, D. Peptidoglycan molecular requirements allowing detection by Nod1 and Nod2. J. Biol. Chem. 2003, 278, 41702–41708. [Google Scholar] [CrossRef] [Green Version]
- Chamaillard, M.; Hashimoto, M.; Horie, Y.; Masumoto, J.; Qiu, S.; Saab, L.; Ogura, Y.; Kawasaki, A.; Fukase, K.; Kusumoto, S.; et al. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat. Immunol. 2003, 4, 702–707. [Google Scholar] [CrossRef]
- Inohara, N.; Koseki, T.; del Peso, L.; Hu, Y.; Yee, C.; Chen, S.; Carrio, R.; Merino, J.; Liu, D.; Ni, J.; et al. Nod1, an Apaf-1-like activator of caspase-9 and nuclear factor-kappaB. J. Biol. Chem. 1999, 274, 14560–14567. [Google Scholar] [CrossRef] [Green Version]
- Ogura, Y.; Inohara, N.; Benito, A.; Chen, F.F.; Yamaoka, S.; Nunez, G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J. Biol. Chem. 2001, 276, 4812–4818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Hara, H.; Nunez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Cooney, R.; Baker, J.; Brain, O.; Danis, B.; Pichulik, T.; Allan, P.; Ferguson, D.J.; Campbell, B.J.; Jewell, D.; Simmons, A. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat. Med. 2010, 16, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Travassos, L.H.; Carneiro, L.A.; Ramjeet, M.; Hussey, S.; Kim, Y.G.; Magalhaes, J.G.; Yuan, L.; Soares, F.; Chea, E.; Le Bourhis, L.; et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 2010, 11, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Denecker, G.; Kalai, M.; D’Hondt, K.; Meeus, A.; Declercq, W.; Saelens, X.; Vandenabeele, P. INCA, a novel human caspase recruitment domain protein that inhibits interleukin-1beta generation. J. Biol. Chem. 2004, 279, 51729–51738. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Stehlik, C.; Reed, J.C. Cop, a caspase recruitment domain-containing protein and inhibitor of caspase-1 activation processing. J. Biol. Chem. 2001, 276, 34495–34500. [Google Scholar] [CrossRef] [Green Version]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Schmidt, T.; Schmid-Burgk, J.L.; Rapino, F.; Robertson, A.A.; Cooper, M.A.; Graf, T.; Hornung, V. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity 2016, 44, 833–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vance, R.E.; Isberg, R.R.; Portnoy, D.A. Patterns of pathogenesis: Discrimination of pathogenic and nonpathogenic microbes by the innate immune system. Cell Host Microbe 2009, 6, 10–21. [Google Scholar] [CrossRef] [Green Version]
- Liston, A.; Masters, S.L. Homeostasis-altering molecular processes as mechanisms of inflammasome activation. Nat. Rev. Immunol. 2017, 17, 208–214. [Google Scholar] [CrossRef]
- Rubartelli, A.; Sitia, R. Stress as an intercellular signal: The emergence of stress-associated molecular patterns (SAMP). Antioxid. Redox Signal. 2009, 11, 2621–2629. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, T.; Dehghani, F. The Cytoskeleton-A Complex Interacting Meshwork. Cells 2019, 8, 362. [Google Scholar] [CrossRef] [Green Version]
- Tapon, N.; Hall, A. Rho, Rac and Cdc42 GTPases regulate the organization of the actin cytoskeleton. Curr. Opin. Cell Biol. 1997, 9, 86–92. [Google Scholar] [CrossRef]
- Sit, S.T.; Manser, E. Rho GTPases and their role in organizing the actin cytoskeleton. J. Cell Sci. 2011, 124, 679–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenny, B. Phosphorylation of tyrosine 474 of the enteropathogenic Escherichia coli (EPEC) Tir receptor molecule is essential for actin nucleating activity and is preceded by additional host modifications. Mol. Microbiol. 1999, 31, 1229–1241. [Google Scholar] [CrossRef]
- Rosenshine, I.; Ruschkowski, S.; Stein, M.; Reinscheid, D.J.; Mills, S.D.; Finlay, B.B. A pathogenic bacterium triggers epithelial signals to form a functional bacterial receptor that mediates actin pseudopod formation. EMBO J. 1996, 15, 2613–2624. [Google Scholar] [CrossRef]
- Kalman, D.; Weiner, O.D.; Goosney, D.L.; Sedat, J.W.; Finlay, B.B.; Abo, A.; Bishop, J.M. Enteropathogenic E. coli acts through WASP and Arp2/3 complex to form actin pedestals. Nat. Cell Biol. 1999, 1, 389–391. [Google Scholar] [CrossRef]
- Alto, N.M.; Weflen, A.W.; Rardin, M.J.; Yarar, D.; Lazar, C.S.; Tonikian, R.; Koller, A.; Taylor, S.S.; Boone, C.; Sidhu, S.S.; et al. The type III effector EspF coordinates membrane trafficking by the spatiotemporal activation of two eukaryotic signaling pathways. J. Cell Biol. 2007, 178, 1265–1278. [Google Scholar] [CrossRef]
- Hardwidge, P.R.; Deng, W.; Vallance, B.A.; Rodriguez-Escudero, I.; Cid, V.J.; Molina, M.; Finlay, B.B. Modulation of host cytoskeleton function by the enteropathogenic Escherichia coli and Citrobacter rodentium effector protein EspG. Infect. Immun. 2005, 73, 2586–2594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Just, I.; Selzer, J.; Wilm, M.; von Eichel-Streiber, C.; Mann, M.; Aktories, K. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature 1995, 375, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Voth, D.E.; Ballard, J.D. Clostridium difficile toxins: Mechanism of action and role in disease. Clin. Microbiol. Rev. 2005, 18, 247–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alto, N.M.; Shao, F.; Lazar, C.S.; Brost, R.L.; Chua, G.; Mattoo, S.; McMahon, S.A.; Ghosh, P.; Hughes, T.R.; Boone, C.; et al. Identification of a bacterial type III effector family with G protein mimicry functions. Cell 2006, 124, 133–145. [Google Scholar] [CrossRef] [Green Version]
- Tran Van Nhieu, G.; Caron, E.; Hall, A.; Sansonetti, P.J. IpaC induces actin polymerization and filopodia formation during Shigella entry into epithelial cells. EMBO J. 1999, 18, 3249–3262. [Google Scholar] [CrossRef] [Green Version]
- Mounier, J.; Popoff, M.R.; Enninga, J.; Frame, M.C.; Sansonetti, P.J.; Van Nhieu, G.T. The IpaC carboxyterminal effector domain mediates Src-dependent actin polymerization during Shigella invasion of epithelial cells. PLoS Pathog. 2009, 5, e1000271. [Google Scholar] [CrossRef] [Green Version]
- Uchiya, K.; Tobe, T.; Komatsu, K.; Suzuki, T.; Watarai, M.; Fukuda, I.; Yoshikawa, M.; Sasakawa, C. Identification of a novel virulence gene, virA, on the large plasmid of Shigella, involved in invasion and intercellular spreading. Mol. Microbiol. 1995, 17, 241–250. [Google Scholar] [CrossRef]
- Yoshida, S.; Katayama, E.; Kuwae, A.; Mimuro, H.; Suzuki, T.; Sasakawa, C. Shigella deliver an effector protein to trigger host microtubule destabilization, which promotes Rac1 activity and efficient bacterial internalization. EMBO J. 2002, 21, 2923–2935. [Google Scholar] [CrossRef]
- Hardt, W.D.; Chen, L.M.; Schuebel, K.E.; Bustelo, X.R.; Galan, J.E.S. S. typhimurium encodes an activator of Rho GTPases that induces membrane ruffling and nuclear responses in host cells. Cell 1998, 93, 815–826. [Google Scholar] [CrossRef] [Green Version]
- Stender, S.; Friebel, A.; Linder, S.; Rohde, M.; Mirold, S.; Hardt, W.D. Identification of SopE2 from Salmonella typhimurium, a conserved guanine nucleotide exchange factor for Cdc42 of the host cell. Mol. Microbiol. 2000, 36, 1206–1221. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Chen, L.M.; Hernandez, L.; Shears, S.B.; Galan, J.E. A Salmonella inositol polyphosphatase acts in conjunction with other bacterial effectors to promote host cell actin cytoskeleton rearrangements and bacterial internalization. Mol. Microbiol. 2001, 39, 248–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGhie, E.J.; Hayward, R.D.; Koronakis, V. Control of actin turnover by a salmonella invasion protein. Mol. Cell 2004, 13, 497–510. [Google Scholar] [CrossRef] [Green Version]
- Jewett, T.J.; Fischer, E.R.; Mead, D.J.; Hackstadt, T. Chlamydial TARP is a bacterial nucleator of actin. Proc. Natl. Acad. Sci. USA 2006, 103, 15599–15604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizarro-Cerda, J.; Kuhbacher, A.; Cossart, P. Entry of Listeria monocytogenes in mammalian epithelial cells: An updated view. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef]
- Van Schaik, E.J.; Chen, C.; Mertens, K.; Weber, M.M.; Samuel, J.E. Molecular pathogenesis of the obligate intracellular bacterium Coxiella burnetii. Nat. Rev. Microbiol. 2013, 11, 561–573. [Google Scholar] [CrossRef] [Green Version]
- Ke, Y.; Chen, Z.; Yang, R. Yersinia pestis: Mechanisms of entry into and resistance to the host cell. Front. Cell. Infect. Microbiol. 2013, 3, 106. [Google Scholar] [CrossRef] [Green Version]
- Colonne, P.M.; Winchell, C.G.; Voth, D.E. Hijacking Host Cell Highways: Manipulation of the Host Actin Cytoskeleton by Obligate Intracellular Bacterial Pathogens. Front. Cell. Infect. Microbiol. 2016, 6, 107. [Google Scholar] [CrossRef] [Green Version]
- Clement, C.; Tiwari, V.; Scanlan, P.M.; Valyi-Nagy, T.; Yue, B.Y.; Shukla, D. A novel role for phagocytosis-like uptake in herpes simplex virus entry. J. Cell Biol. 2006, 174, 1009–1021. [Google Scholar] [CrossRef]
- Harmon, B.; Campbell, N.; Ratner, L. Role of Abl kinase and the Wave2 signaling complex in HIV-1 entry at a post-hemifusion step. PLoS Pathog. 2010, 6, e1000956. [Google Scholar] [CrossRef] [Green Version]
- Chandra, S.; Kalaivani, R.; Kumar, M.; Srinivasan, N.; Sarkar, D.P. Sendai virus recruits cellular villin to remodel actin cytoskeleton during fusion with hepatocytes. Mol. Biol. Cell 2017, 28, 3801–3814. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.L.; Lidke, D.S.; Ozbun, M.A. Virus activated filopodia promote human papillomavirus type 31 uptake from the extracellular matrix. Virology 2008, 381, 16–21. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, V.; Combe, A.; David, V.; Malmquist, N.A.; Delorme, V.; Leroy, C.; Blazquez, S.; Menard, R.; Tardieux, I. Host cell entry by apicomplexa parasites requires actin polymerization in the host cell. Cell Host Microbe 2009, 5, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Delorme-Walker, V.; Abrivard, M.; Lagal, V.; Anderson, K.; Perazzi, A.; Gonzalez, V.; Page, C.; Chauvet, J.; Ochoa, W.; Volkmann, N.; et al. Toxofilin upregulates the host cortical actin cytoskeleton dynamics, facilitating Toxoplasma invasion. J. Cell Sci. 2012, 125, 4333–4342. [Google Scholar] [CrossRef] [Green Version]
- Roggwiller, E.; Betoulle, M.E.; Blisnick, T.; Braun Breton, C. A role for erythrocyte band 3 degradation by the parasite gp76 serine protease in the formation of the parasitophorous vacuole during invasion of erythrocytes by Plasmodium falciparum. Mol. Biochem. Parasitol. 1996, 82, 13–24. [Google Scholar] [CrossRef]
- McPherson, R.A.; Donald, D.R.; Sawyer, W.H.; Tilley, L. Proteolytic digestion of band 3 at an external site alters the erythrocyte membrane organisation and may facilitate malarial invasion. Mol. Biochem. Parasitol. 1993, 62, 233–242. [Google Scholar] [CrossRef]
- Le Bonniec, S.; Deregnaucourt, C.; Redeker, V.; Banerjee, R.; Grellier, P.; Goldberg, D.E.; Schrevel, J. Plasmepsin II, an acidic hemoglobinase from the Plasmodium falciparum food vacuole, is active at neutral pH on the host erythrocyte membrane skeleton. J. Biol. Chem. 1999, 274, 14218–14223. [Google Scholar] [CrossRef] [Green Version]
- Raphael, P.; Takakuwa, Y.; Manno, S.; Liu, S.C.; Chishti, A.H.; Hanspal, M. A cysteine protease activity from Plasmodium falciparum cleaves human erythrocyte ankyrin. Mol. Biochem. Parasitol. 2000, 110, 259–272. [Google Scholar] [CrossRef]
- Ehsani, S.; Santos, J.C.; Rodrigues, C.D.; Henriques, R.; Audry, L.; Zimmer, C.; Sansonetti, P.; Tran Van Nhieu, G.; Enninga, J. Hierarchies of host factor dynamics at the entry site of Shigella flexneri during host cell invasion. Infect. Immun. 2012, 80, 2548–2557. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, S.; Bergqvist, J.; Gil, M.; Valenzuela, C.; Barrio, L.; Lebreton, S.; Zurzolo, C.; Enninga, J. Actin Assembly around the Shigella-Containing Vacuole Promotes Successful Infection. Cell Rep. 2020, 31, 107638. [Google Scholar] [CrossRef]
- Kumar, Y.; Valdivia, R.H. Actin and intermediate filaments stabilize the Chlamydia trachomatis vacuole by forming dynamic structural scaffolds. Cell Host Microbe 2008, 4, 159–169. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, M.; Salinas, R.; Rosales, E.; Carminati, S.; Colombo, M.I.; Beron, W. Actin dynamics and Rho GTPases regulate the size and formation of parasitophorous vacuoles containing Coxiella burnetii. Infect. Immun. 2009, 77, 4609–4620. [Google Scholar] [CrossRef] [Green Version]
- Kocks, C.; Gouin, E.; Tabouret, M.; Berche, P.; Ohayon, H.; Cossart, P.L. monocytogenes-induced actin assembly requires the actA gene product, a surface protein. Cell 1992, 68, 521–531. [Google Scholar] [CrossRef]
- Boujemaa-Paterski, R.; Gouin, E.; Hansen, G.; Samarin, S.; Le Clainche, C.; Didry, D.; Dehoux, P.; Cossart, P.; Kocks, C.; Carlier, M.F.; et al. Listeria protein ActA mimics WASp family proteins: It activates filament barbed end branching by Arp2/3 complex. Biochemistry 2001, 40, 11390–11404. [Google Scholar] [CrossRef]
- Zalevsky, J.; Grigorova, I.; Mullins, R.D. Activation of the Arp2/3 complex by the Listeria acta protein. Acta binds two actin monomers and three subunits of the Arp2/3 complex. J. Biol. Chem. 2001, 276, 3468–3475. [Google Scholar] [CrossRef] [Green Version]
- Makino, S.; Sasakawa, C.; Kamata, K.; Kurata, T.; Yoshikawa, M. A genetic determinant required for continuous reinfection of adjacent cells on large plasmid in S. flexneri 2a. Cell 1986, 46, 551–555. [Google Scholar] [CrossRef]
- Bernardini, M.L.; Mounier, J.; d’Hauteville, H.; Coquis-Rondon, M.; Sansonetti, P.J. Identification of icsA, a plasmid locus of Shigella flexneri that governs bacterial intra- and intercellular spread through interaction with F-actin. Proc. Natl. Acad. Sci. USA 1989, 86, 3867–3871. [Google Scholar] [CrossRef] [Green Version]
- Egile, C.; Loisel, T.P.; Laurent, V.; Li, R.; Pantaloni, D.; Sansonetti, P.J.; Carlier, M.F. Activation of the CDC42 effector N-WASP by the Shigella flexneri IcsA protein promotes actin nucleation by Arp2/3 complex and bacterial actin-based motility. J. Cell Biol. 1999, 146, 1319–1332. [Google Scholar] [CrossRef] [Green Version]
- Reed, S.C.O.; Lamason, R.L.; Risca, V.I.; Abernathy, E.; Welch, M.D. Rickettsia actin-based motility occurs in distinct phases mediated by different actin nucleators. Curr. Biol. 2014, 24, 98–103. [Google Scholar] [CrossRef] [Green Version]
- Benanti, E.L.; Nguyen, C.M.; Welch, M.D. Virulent Burkholderia species mimic host actin polymerases to drive actin-based motility. Cell 2015, 161, 348–360. [Google Scholar] [CrossRef] [Green Version]
- Dabiri, G.A.; Sanger, J.M.; Portnoy, D.A.; Southwick, F.S. Listeria monocytogenes moves rapidly through the host-cell cytoplasm by inducing directional actin assembly. Proc. Natl. Acad. Sci. USA 1990, 87, 6068–6072. [Google Scholar] [CrossRef] [Green Version]
- Cudmore, S.; Cossart, P.; Griffiths, G.; Way, M. Actin-based motility of vaccinia virus. Nature 1995, 378, 636–638. [Google Scholar] [CrossRef]
- Scaplehorn, N.; Holmstrom, A.; Moreau, V.; Frischknecht, F.; Reckmann, I.; Way, M. Grb2 and Nck act cooperatively to promote actin-based motility of vaccinia virus. Curr. Biol. 2002, 12, 740–745. [Google Scholar] [CrossRef] [Green Version]
- Ohkawa, T.; Volkman, L.E.; Welch, M.D. Actin-based motility drives baculovirus transit to the nucleus and cell surface. J. Cell Biol. 2010, 190, 187–195. [Google Scholar] [CrossRef] [Green Version]
- Schudt, G.; Dolnik, O.; Kolesnikova, L.; Biedenkopf, N.; Herwig, A.; Becker, S. Transport of Ebolavirus Nucleocapsids Is Dependent on Actin Polymerization: Live-Cell Imaging Analysis of Ebolavirus-Infected Cells. J. Infect. Dis. 2015, 212 (Suppl. 2), S160–S166. [Google Scholar] [CrossRef]
- Favoreel, H.W.; Van Minnebruggen, G.; Adriaensen, D.; Nauwynck, H.J. Cytoskeletal rearrangements and cell extensions induced by the US3 kinase of an alphaherpesvirus are associated with enhanced spread. Proc. Natl. Acad. Sci. USA 2005, 102, 8990–8995. [Google Scholar] [CrossRef] [Green Version]
- Dixit, R.; Tiwari, V.; Shukla, D. Herpes simplex virus type 1 induces filopodia in differentiated P19 neural cells to facilitate viral spread. Neurosci. Lett. 2008, 440, 113–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Najjar, F.; Cifuentes-Munoz, N.; Chen, J.; Zhu, H.; Buchholz, U.J.; Moncman, C.L.; Dutch, R.E. Human metapneumovirus Induces Reorganization of the Actin Cytoskeleton for Direct Cell-to-Cell Spread. PLoS Pathog. 2016, 12, e1005922. [Google Scholar] [CrossRef] [Green Version]
- Schroder, M.; Kaufman, R.J. ER stress and the unfolded protein response. Mutat. Res. 2005, 569, 29–63. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef]
- Li, S.; Kong, L.; Yu, X. The expanding roles of endoplasmic reticulum stress in virus replication and pathogenesis. Crit. Rev. Microbiol. 2015, 41, 150–164. [Google Scholar] [CrossRef]
- Galindo, I.; Hernaez, B.; Munoz-Moreno, R.; Cuesta-Geijo, M.A.; Dalmau-Mena, I.; Alonso, C. The ATF6 branch of unfolded protein response and apoptosis are activated to promote African swine fever virus infection. Cell Death Dis. 2012, 3, e341. [Google Scholar] [CrossRef] [Green Version]
- Andres, G.; Garcia-Escudero, R.; Simon-Mateo, C.; Vinuela, E. African swine fever virus is enveloped by a two-membraned collapsed cisterna derived from the endoplasmic reticulum. J. Virol. 1998, 72, 8988–9001. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.P.; Siu, K.L.; Chin, K.T.; Yuen, K.Y.; Zheng, B.; Jin, D.Y. Modulation of the unfolded protein response by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2006, 80, 9279–9287. [Google Scholar] [CrossRef] [Green Version]
- Minakshi, R.; Padhan, K.; Rani, M.; Khan, N.; Ahmad, F.; Jameel, S. The SARS Coronavirus 3a protein causes endoplasmic reticulum stress and induces ligand-independent downregulation of the type 1 interferon receptor. PLoS ONE 2009, 4, e8342. [Google Scholar] [CrossRef]
- Sung, S.C.; Chao, C.Y.; Jeng, K.S.; Yang, J.Y.; Lai, M.M. The 8ab protein of SARS-CoV is a luminal ER membrane-associated protein and induces the activation of ATF6. Virology 2009, 387, 402–413. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.Y.; Hsu, Y.W.; Liao, C.L.; Lin, Y.L. Flavivirus infection activates the XBP1 pathway of the unfolded protein response to cope with endoplasmic reticulum stress. J. Virol. 2006, 80, 11868–11880. [Google Scholar] [CrossRef] [Green Version]
- Umareddy, I.; Pluquet, O.; Wang, Q.Y.; Vasudevan, S.G.; Chevet, E.; Gu, F. Dengue virus serotype infection specifies the activation of the unfolded protein response. Virol. J. 2007, 4, 91. [Google Scholar] [CrossRef] [Green Version]
- Limjindaporn, T.; Wongwiwat, W.; Noisakran, S.; Srisawat, C.; Netsawang, J.; Puttikhunt, C.; Kasinrerk, W.; Avirutnan, P.; Thiemmeca, S.; Sriburi, R.; et al. Interaction of dengue virus envelope protein with endoplasmic reticulum-resident chaperones facilitates dengue virus production. Biochem. Biophys. Res. Commun. 2009, 379, 196–200. [Google Scholar] [CrossRef]
- Pena, J.; Harris, E. Dengue virus modulates the unfolded protein response in a time-dependent manner. J. Biol. Chem. 2011, 286, 14226–14236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.H.; Shin, D.M.; Kang, G.; Kim, K.H.; Park, J.B.; Hur, G.M.; Lee, H.M.; Lim, Y.J.; Park, J.K.; Jo, E.K.; et al. Endoplasmic reticulum stress response is involved in Mycobacterium tuberculosis protein ESAT-6-mediated apoptosis. FEBS Lett. 2010, 584, 2445–2454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.A.; Lim, Y.J.; Cho, S.N.; Lee, J.H.; Jeong, J.A.; Kim, E.J.; Park, J.B.; Kim, S.H.; Park, H.S.; Kim, H.J.; et al. Mycobacterial HBHA induces endoplasmic reticulum stress-mediated apoptosis through the generation of reactive oxygen species and cytosolic Ca2+ in murine macrophage RAW 264.7 cells. Cell Death Dis. 2013, 4, e957. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y.; Lee, M.S.; Cherla, R.P.; Tesh, V.L. Shiga toxin 1 induces apoptosis through the endoplasmic reticulum stress response in human monocytic cells. Cell Microbiol. 2008, 10, 770–780. [Google Scholar] [CrossRef]
- Pillich, H.; Loose, M.; Zimmer, K.P.; Chakraborty, T. Activation of the unfolded protein response by Listeria monocytogenes. Cell Microbiol. 2012, 14, 949–964. [Google Scholar] [CrossRef] [PubMed]
- George, Z.; Omosun, Y.; Azenabor, A.A.; Partin, J.; Joseph, K.; Ellerson, D.; He, Q.; Eko, F.; Bandea, C.; Svoboda, P.; et al. The Roles of Unfolded Protein Response Pathways in Chlamydia Pathogenesis. J. Infect. Dis. 2017, 215, 456–465. [Google Scholar] [CrossRef] [Green Version]
- Pham, O.H.; Lee, B.; Labuda, J.; Keestra-Gounder, A.M.; Byndloss, M.X.; Tsolis, R.M.; McSorley, S.J. NOD1/NOD2 and RIP2 Regulate Endoplasmic Reticulum Stress-Induced Inflammation during Chlamydia Infection. mBio 2020, 11. [Google Scholar] [CrossRef]
- De Jong, M.F.; Starr, T.; Winter, M.G.; den Hartigh, A.B.; Child, R.; Knodler, L.A.; van Dijl, J.M.; Celli, J.; Tsolis, R.M. Sensing of bacterial type IV secretion via the unfolded protein response. mBio 2013, 4, e00418-12. [Google Scholar] [CrossRef] [Green Version]
- Guimaraes, E.S.; Gomes, M.T.R.; Campos, P.C.; Mansur, D.S.; Dos Santos, A.A.; Harms, J.; Splitter, G.; Smith, J.A.; Barber, G.N.; Oliveira, S.C. Brucella abortus Cyclic Dinucleotides Trigger STING-Dependent Unfolded Protein Response That Favors Bacterial Replication. J. Immunol. 2019, 202, 2671–2681. [Google Scholar] [CrossRef]
- Vajjala, A.; Biswas, D.; Tay, W.H.; Hanski, E.; Kline, K.A. Streptolysin-induced endoplasmic reticulum stress promotes group A Streptococcal host-associated biofilm formation and necrotising fasciitis. Cell Microbiol. 2019, 21, e12956. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, A.; Beare, P.A.; Schulze-Luehrmann, J.; Cordsmeier, A.; Pazen, T.; Sonnewald, S.; Luhrmann, A. The Coxiella burnetii effector protein CaeB modulates endoplasmatic reticulum (ER) stress signalling and is required for efficient replication in Galleria mellonella. Cell Microbiol. 2021, 23, e13305. [Google Scholar] [CrossRef]
- Inacio, P.; Zuzarte-Luis, V.; Ruivo, M.T.; Falkard, B.; Nagaraj, N.; Rooijers, K.; Mann, M.; Mair, G.; Fidock, D.A.; Mota, M.M. Parasite-induced ER stress response in hepatocytes facilitates Plasmodium liver stage infection. EMBO Rep. 2015, 16, 955–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augusto, L.; Martynowicz, J.; Amin, P.H.; Alakhras, N.S.; Kaplan, M.H.; Wek, R.C.; Sullivan, W.J., Jr. Toxoplasma gondii Co-opts the Unfolded Protein Response To Enhance Migration and Dissemination of Infected Host Cells. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Shadel, G.S.; Ghosh, S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011, 11, 389–402. [Google Scholar] [CrossRef] [Green Version]
- Tiku, V.; Tan, M.W.; Dikic, I. Mitochondrial Functions in Infection and Immunity. Trends Cell Biol. 2020, 30, 263–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [Green Version]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef]
- Xu, L.G.; Wang, Y.Y.; Han, K.J.; Li, L.Y.; Zhai, Z.; Shu, H.B. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 2005, 19, 727–740. [Google Scholar] [CrossRef]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef] [Green Version]
- Abuaita, B.H.; Schultz, T.L.; O’Riordan, M.X. Mitochondria-Derived Vesicles Deliver Antimicrobial Reactive Oxygen Species to Control Phagosome-Localized Staphylococcus aureus. Cell Host Microbe 2018, 24, 625–636. [Google Scholar] [CrossRef] [Green Version]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Pearce, E.J. Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 2016, 213, 15–23. [Google Scholar] [CrossRef]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Bambouskova, M.; Gorvel, L.; Lampropoulou, V.; Sergushichev, A.; Loginicheva, E.; Johnson, K.; Korenfeld, D.; Mathyer, M.E.; Kim, H.; Huang, L.H.; et al. Electrophilic properties of itaconate and derivatives regulate the IkappaBzeta-ATF3 inflammatory axis. Nature 2018, 556, 501–504. [Google Scholar] [CrossRef]
- Stavru, F.; Bouillaud, F.; Sartori, A.; Ricquier, D.; Cossart, P. Listeria monocytogenes transiently alters mitochondrial dynamics during infection. Proc. Natl. Acad. Sci. USA 2011, 108, 3612–3617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, F.; Spier, A.; Chaze, T.; Matondo, M.; Cossart, P.; Stavru, F. Listeria monocytogenes Exploits Mitochondrial Contact Site and Cristae Organizing System Complex Subunit Mic10 to Promote Mitochondrial Fragmentation and Cellular Infection. mBio 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Stavru, F.; Palmer, A.E.; Wang, C.; Youle, R.J.; Cossart, P. Atypical mitochondrial fission upon bacterial infection. Proc. Natl. Acad. Sci. USA 2013, 110, 16003–16008. [Google Scholar] [CrossRef] [Green Version]
- Lobet, E.; Willemart, K.; Ninane, N.; Demazy, C.; Sedzicki, J.; Lelubre, C.; De Bolle, X.; Renard, P.; Raes, M.; Dehio, C.; et al. Mitochondrial fragmentation affects neither the sensitivity to TNFalpha-induced apoptosis of Brucella-infected cells nor the intracellular replication of the bacteria. Sci. Rep. 2018, 8, 5173. [Google Scholar] [CrossRef] [Green Version]
- Jain, P.; Luo, Z.Q.; Blanke, S.R. Helicobacter pylori vacuolating cytotoxin A (VacA) engages the mitochondrial fission machinery to induce host cell death. Proc. Natl. Acad. Sci. USA 2011, 108, 16032–16037. [Google Scholar] [CrossRef] [Green Version]
- Lum, M.; Morona, R. Dynamin-related protein Drp1 and mitochondria are important for Shigella flexneri infection. Int. J. Med. Microbiol. 2014, 304, 530–541. [Google Scholar] [CrossRef] [Green Version]
- Escoll, P.; Song, O.R.; Viana, F.; Steiner, B.; Lagache, T.; Olivo-Marin, J.C.; Impens, F.; Brodin, P.; Hilbi, H.; Buchrieser, C. Legionella pneumophila Modulates Mitochondrial Dynamics to Trigger Metabolic Repurposing of Infected Macrophages. Cell Host Microbe 2017, 22, 302–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshizumi, T.; Ichinohe, T.; Sasaki, O.; Otera, H.; Kawabata, S.; Mihara, K.; Koshiba, T. Influenza A virus protein PB1-F2 translocates into mitochondria via Tom40 channels and impairs innate immunity. Nat. Commun. 2014, 5, 4713. [Google Scholar] [CrossRef]
- Shi, C.S.; Qi, H.Y.; Boularan, C.; Huang, N.N.; Abu-Asab, M.; Shelhamer, J.H.; Kehrl, J.H. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 2014, 193, 3080–3089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fields, J.A.; Serger, E.; Campos, S.; Divakaruni, A.S.; Kim, C.; Smith, K.; Trejo, M.; Adame, A.; Spencer, B.; Rockenstein, E.; et al. HIV alters neuronal mitochondrial fission/fusion in the brain during HIV-associated neurocognitive disorders. Neurobiol. Dis. 2016, 86, 154–169. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Salamon, H.; Eugenin, E.A.; Pine, R.; Cooper, A.; Gennaro, M.L. Infection with Mycobacterium tuberculosis induces the Warburg effect in mouse lungs. Sci. Rep. 2015, 5, 18176. [Google Scholar] [CrossRef]
- Gleeson, L.E.; Sheedy, F.J.; Palsson-McDermott, E.M.; Triglia, D.; O’Leary, S.M.; O’Sullivan, M.P.; O’Neill, L.A.; Keane, J. Cutting Edge: Mycobacterium tuberculosis Induces Aerobic Glycolysis in Human Alveolar Macrophages That Is Required for Control of Intracellular Bacillary Replication. J. Immunol. 2016, 196, 2444–2449. [Google Scholar] [CrossRef] [Green Version]
- Lachmandas, E.; Beigier-Bompadre, M.; Cheng, S.C.; Kumar, V.; van Laarhoven, A.; Wang, X.; Ammerdorffer, A.; Boutens, L.; de Jong, D.; Kanneganti, T.D.; et al. Rewiring cellular metabolism via the AKT/mTOR pathway contributes to host defence against Mycobacterium tuberculosis in human and murine cells. Eur. J. Immunol. 2016, 46, 2574–2586. [Google Scholar] [CrossRef]
- Hackett, E.E.; Charles-Messance, H.; O’Leary, S.M.; Gleeson, L.E.; Munoz-Wolf, N.; Case, S.; Wedderburn, A.; Johnston, D.G.W.; Williams, M.A.; Smyth, A.; et al. Mycobacterium tuberculosis Limits Host Glycolysis and IL-1beta by Restriction of PFK-M via MicroRNA-21. Cell Rep. 2020, 30, 124–136. [Google Scholar] [CrossRef]
- Cumming, B.M.; Addicott, K.W.; Adamson, J.H.; Steyn, A.J. Mycobacterium tuberculosis induces decelerated bioenergetic metabolism in human macrophages. eLife 2018, 7. [Google Scholar] [CrossRef]
- Czyz, D.M.; Willett, J.W.; Crosson, S. Brucella abortus Induces a Warburg Shift in Host Metabolism That Is Linked to Enhanced Intracellular Survival of the Pathogen. J. Bacteriol. 2017, 199. [Google Scholar] [CrossRef] [Green Version]
- Kentner, D.; Martano, G.; Callon, M.; Chiquet, P.; Brodmann, M.; Burton, O.; Wahlander, A.; Nanni, P.; Delmotte, N.; Grossmann, J.; et al. Shigella reroutes host cell central metabolism to obtain high-flux nutrient supply for vigorous intracellular growth. Proc. Natl. Acad. Sci. USA 2014, 111, 9929–9934. [Google Scholar] [CrossRef] [Green Version]
- Metheni, M.; Lombes, A.; Bouillaud, F.; Batteux, F.; Langsley, G. HIF-1alpha induction, proliferation and glycolysis of Theileria-infected leukocytes. Cell. Microbiol. 2015, 17, 467–472. [Google Scholar] [CrossRef]
- Medjkane, S.; Perichon, M.; Marsolier, J.; Dairou, J.; Weitzman, J.B. Theileria induces oxidative stress and HIF1alpha activation that are essential for host leukocyte transformation. Oncogene 2014, 33, 1809–1817. [Google Scholar] [CrossRef]
- Marsolier, J.; Perichon, M.; Weitzman, J.B.; Medjkane, S. Secreted parasite Pin1 isomerase stabilizes host PKM2 to reprogram host cell metabolism. Commun. Biol. 2019, 2, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hargrave, K.E.; Woods, S.; Millington, O.; Chalmers, S.; Westrop, G.D.; Roberts, C.W. Multi-Omics Studies Demonstrate Toxoplasma gondii-Induced Metabolic Reprogramming of Murine Dendritic Cells. Front. Cell. Infect. Microbiol. 2019, 9, 309. [Google Scholar] [CrossRef]
- Olson, W.J.; Martorelli Di Genova, B.; Gallego-Lopez, G.; Dawson, A.R.; Stevenson, D.; Amador-Noguez, D.; Knoll, L.J. Dual metabolomic profiling uncovers Toxoplasma manipulation of the host metabolome and the discovery of a novel parasite metabolic capability. PLoS Pathog. 2020, 16, e1008432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blader, I.J.; Manger, I.D.; Boothroyd, J.C. Microarray analysis reveals previously unknown changes in Toxoplasma gondii-infected human cells. J. Biol. Chem. 2001, 276, 24223–24231. [Google Scholar] [CrossRef] [Green Version]
- Shah-Simpson, S.; Lentini, G.; Dumoulin, P.C.; Burleigh, B.A. Modulation of host central carbon metabolism and in situ glucose uptake by intracellular Trypanosoma cruzi amastigotes. PLoS Pathog. 2017, 13, e1006747. [Google Scholar] [CrossRef] [Green Version]
- Wallqvist, A.; Fang, X.; Tewari, S.G.; Ye, P.; Reifman, J. Metabolic host responses to malarial infection during the intraerythrocytic developmental cycle. BMC Syst. Biol. 2016, 10, 58. [Google Scholar] [CrossRef] [Green Version]
- Vastag, L.; Koyuncu, E.; Grady, S.L.; Shenk, T.E.; Rabinowitz, J.D. Divergent effects of human cytomegalovirus and herpes simplex virus-1 on cellular metabolism. PLoS Pathog. 2011, 7, e1002124. [Google Scholar] [CrossRef] [Green Version]
- Diamond, D.L.; Syder, A.J.; Jacobs, J.M.; Sorensen, C.M.; Walters, K.A.; Proll, S.C.; McDermott, J.E.; Gritsenko, M.A.; Zhang, Q.; Zhao, R.; et al. Temporal proteome and lipidome profiles reveal hepatitis C virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog. 2010, 6, e1000719. [Google Scholar] [CrossRef] [Green Version]
- Mohr, I.; Sonenberg, N. Host translation at the nexus of infection and immunity. Cell Host Microbe 2012, 12, 470–483. [Google Scholar] [CrossRef] [Green Version]
- Walsh, D.; Mathews, M.B.; Mohr, I. Tinkering with translation: Protein synthesis in virus-infected cells. Cold Spring Harb. Perspect. Biol. 2013, 5, a012351. [Google Scholar] [CrossRef]
- Jackson, R.J.; Hellen, C.U.; Pestova, T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etchison, D.; Milburn, S.C.; Edery, I.; Sonenberg, N.; Hershey, J.W. Inhibition of HeLa cell protein synthesis following poliovirus infection correlates with the proteolysis of a 220,000-dalton polypeptide associated with eucaryotic initiation factor 3 and a cap binding protein complex. J. Biol. Chem. 1982, 257, 14806–14810. [Google Scholar] [CrossRef]
- Lamphear, B.J.; Yan, R.; Yang, F.; Waters, D.; Liebig, H.D.; Klump, H.; Kuechler, E.; Skern, T.; Rhoads, R.E. Mapping the cleavage site in protein synthesis initiation factor eIF-4 gamma of the 2A proteases from human Coxsackievirus and rhinovirus. J. Biol. Chem. 1993, 268, 19200–19203. [Google Scholar] [CrossRef]
- Ventoso, I.; Blanco, R.; Perales, C.; Carrasco, L. HIV-1 protease cleaves eukaryotic initiation factor 4G and inhibits cap-dependent translation. Proc. Natl. Acad. Sci. USA 2001, 98, 12966–12971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, E.; Menendez-Arias, L.; Carrasco, L. The eukaryotic translation initiation factor 4GI is cleaved by different retroviral proteases. J. Virol. 2003, 77, 12392–12400. [Google Scholar] [CrossRef] [Green Version]
- Willcocks, M.M.; Carter, M.J.; Roberts, L.O. Cleavage of eukaryotic initiation factor eIF4G and inhibition of host-cell protein synthesis during feline calicivirus infection. J. Gen. Virol. 2004, 85, 1125–1130. [Google Scholar] [CrossRef]
- Gradi, A.; Foeger, N.; Strong, R.; Svitkin, Y.V.; Sonenberg, N.; Skern, T.; Belsham, G.J. Cleavage of eukaryotic translation initiation factor 4GII within foot-and-mouth disease virus-infected cells: Identification of the L-protease cleavage site in vitro. J. Virol. 2004, 78, 3271–3278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piron, M.; Vende, P.; Cohen, J.; Poncet, D. Rotavirus RNA-binding protein NSP3 interacts with eIF4GI and evicts the poly(A) binding protein from eIF4F. EMBO J. 1998, 17, 5811–5821. [Google Scholar] [CrossRef] [Green Version]
- Ho, B.C.; Yu, S.L.; Chen, J.J.; Chang, S.Y.; Yan, B.S.; Hong, Q.S.; Singh, S.; Kao, C.L.; Chen, H.Y.; Su, K.Y.; et al. Enterovirus-induced miR-141 contributes to shutoff of host protein translation by targeting the translation initiation factor eIF4E. Cell Host Microbe 2011, 9, 58–69. [Google Scholar] [CrossRef] [Green Version]
- Thoms, M.; Buschauer, R.; Ameismeier, M.; Koepke, L.; Denk, T.; Hirschenberger, M.; Kratzat, H.; Hayn, M.; Mackens-Kiani, T.; Cheng, J.; et al. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science 2020, 369, 1249–1255. [Google Scholar] [CrossRef]
- Deng, Q.; Barbieri, J.T. Molecular mechanisms of the cytotoxicity of ADP-ribosylating toxins. Annu. Rev. Microbiol. 2008, 62, 271–288. [Google Scholar] [CrossRef]
- Ohmer, M.; Tzivelekidis, T.; Niedenfuhr, N.; Volceanov-Hahn, L.; Barth, S.; Vier, J.; Borries, M.; Busch, H.; Kook, L.; Biniossek, M.L.; et al. Infection of HeLa cells with Chlamydia trachomatis inhibits protein synthesis and causes multiple changes to host cell pathways. Cell Microbiol. 2019, 21, e12993. [Google Scholar] [CrossRef]
- Belyi, Y.; Niggeweg, R.; Opitz, B.; Vogelsgesang, M.; Hippenstiel, S.; Wilm, M.; Aktories, K. Legionella pneumophila glucosyltransferase inhibits host elongation factor 1A. Proc. Natl. Acad. Sci. USA 2006, 103, 16953–16958. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Banga, S.; Liu, Y.; Xu, L.; Gao, P.; Shamovsky, I.; Nudler, E.; Luo, Z.Q. Targeting eEF1A by a Legionella pneumophila effector leads to inhibition of protein synthesis and induction of host stress response. Cell Microbiol. 2009, 11, 911–926. [Google Scholar] [CrossRef] [Green Version]
- Nelson, R.J.; Ziegelhoffer, T.; Nicolet, C.; Werner-Washburne, M.; Craig, E.A. The translation machinery and 70 kd heat shock protein cooperate in protein synthesis. Cell 1992, 71, 97–105. [Google Scholar] [CrossRef]
- Moss, S.M.; Taylor, I.R.; Ruggero, D.; Gestwicki, J.E.; Shokat, K.M.; Mukherjee, S. A Legionella pneumophila Kinase Phosphorylates the Hsp70 Chaperone Family to Inhibit Eukaryotic Protein Synthesis. Cell Host Microbe 2019, 25, 454–462. [Google Scholar] [CrossRef] [Green Version]
- Keestra-Gounder, A.M.; Byndloss, M.X.; Seyffert, N.; Young, B.M.; Chavez-Arroyo, A.; Tsai, A.Y.; Cevallos, S.A.; Winter, M.G.; Pham, O.H.; Tiffany, C.R.; et al. NOD1 and NOD2 signalling links ER stress with inflammation. Nature 2016, 532, 394–397. [Google Scholar] [CrossRef] [Green Version]
- Sandstrom, A.; Mitchell, P.S.; Goers, L.; Mu, E.W.; Lesser, C.F.; Vance, R.E. Functional degradation: A mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science 2019, 364. [Google Scholar] [CrossRef]
- Robinson, K.S.; Teo, D.E.T.; Tan, K.S.; Toh, G.A.; Ong, H.H.; Lim, C.K.; Lay, K.; Au, B.V.; Lew, T.S.; Chu, J.J.H.; et al. Enteroviral 3C protease activates the human NLRP1 inflammasome in airway epithelia. Science 2020, 370. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Legrand-Poels, S.; Kustermans, G.; Bex, F.; Kremmer, E.; Kufer, T.A.; Piette, J. Modulation of Nod2-dependent NF-kappaB signaling by the actin cytoskeleton. J. Cell Sci. 2007, 120, 1299–1310. [Google Scholar] [CrossRef] [Green Version]
- Kustermans, G.; El Benna, J.; Piette, J.; Legrand-Poels, S. Perturbation of actin dynamics induces NF-kappaB activation in myelomonocytic cells through an NADPH oxidase-dependent pathway. Biochem. J. 2005, 387, 531–540. [Google Scholar] [CrossRef]
- Keestra, A.M.; Winter, M.G.; Klein-Douwel, D.; Xavier, M.N.; Winter, S.E.; Kim, A.; Tsolis, R.M.; Baumler, A.J. A Salmonella virulence factor activates the NOD1/NOD2 signaling pathway. mBio 2011, 2. [Google Scholar] [CrossRef] [Green Version]
- Keestra, A.M.; Winter, M.G.; Auburger, J.J.; Frassle, S.P.; Xavier, M.N.; Winter, S.E.; Kim, A.; Poon, V.; Ravesloot, M.M.; Waldenmaier, J.F.; et al. Manipulation of small Rho GTPases is a pathogen-induced process detected by NOD1. Nature 2013, 496, 233–237. [Google Scholar] [CrossRef] [Green Version]
- Nemeth, Z.H.; Deitch, E.A.; Davidson, M.T.; Szabo, C.; Vizi, E.S.; Hasko, G. Disruption of the actin cytoskeleton results in nuclear factor-kappaB activation and inflammatory mediator production in cultured human intestinal epithelial cells. J. Cell. Physiol. 2004, 200, 71–81. [Google Scholar] [CrossRef]
- Eitel, J.; Krull, M.; Hocke, A.C.; N’Guessan, P.D.; Zahlten, J.; Schmeck, B.; Slevogt, H.; Hippenstiel, S.; Suttorp, N.; Opitz, B. Beta-PIX and Rac1 GTPase mediate trafficking and negative regulation of NOD2. J. Immunol. 2008, 181, 2664–2671. [Google Scholar] [CrossRef] [Green Version]
- Brawn, L.C.; Hayward, R.D.; Koronakis, V. Salmonella SPI1 effector SipA persists after entry and cooperates with a SPI2 effector to regulate phagosome maturation and intracellular replication. Cell Host Microbe 2007, 1, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Krendel, M.; Zenke, F.T.; Bokoch, G.M. Nucleotide exchange factor GEF-H1 mediates cross-talk between microtubules and the actin cytoskeleton. Nat. Cell Biol. 2002, 4, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Fukazawa, A.; Alonso, C.; Kurachi, K.; Gupta, S.; Lesser, C.F.; McCormick, B.A.; Reinecker, H.C. GEF-H1 mediated control of NOD1 dependent NF-kappaB activation by Shigella effectors. PLoS Pathog. 2008, 4, e1000228. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Alonso, C.; Ballester, I.; Song, J.H.; Chang, S.Y.; Guleng, B.; Arihiro, S.; Murray, P.J.; Xavier, R.; Kobayashi, K.S.; et al. Control of NOD2 and Rip2-dependent innate immune activation by GEF-H1. Inflamm. Bowel Dis. 2012, 18, 603–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielig, H.; Lautz, K.; Braun, P.R.; Menning, M.; Machuy, N.; Brugmann, C.; Barisic, S.; Eisler, S.A.; Andree, M.; Zurek, B.; et al. The cofilin phosphatase slingshot homolog 1 (SSH1) links NOD1 signaling to actin remodeling. PLoS Pathog. 2014, 10, e1004351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pahl, H.L.; Baeuerle, P.A. A novel signal transduction pathway from the endoplasmic reticulum to the nucleus is mediated by transcription factor NF-kappa B. EMBO J. 1995, 14, 2580–2588. [Google Scholar] [CrossRef]
- Mendez, J.M.; Kolora, L.D.; Lemon, J.S.; Dupree, S.L.; Keestra-Gounder, A.M. Activation of the Endoplasmic Reticulum Stress Response Impacts the NOD1 Signaling Pathway. Infect. Immun. 2019, 87. [Google Scholar] [CrossRef] [Green Version]
- Keestra-Gounder, A.M.; Tsolis, R.M. NOD1 and NOD2: Beyond Peptidoglycan Sensing. Trends Immunol. 2017, 38, 758–767. [Google Scholar] [CrossRef]
- Ozcan, L.; Tabas, I. Role of endoplasmic reticulum stress in metabolic disease and other disorders. Annu. Rev. Med. 2012, 63, 317–328. [Google Scholar] [CrossRef] [Green Version]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [Green Version]
- Molinaro, R.; Mukherjee, T.; Flick, R.; Philpott, D.J.; Girardin, S.E. Trace levels of peptidoglycan in serum underlie the NOD-dependent cytokine response to endoplasmic reticulum stress. J. Biol. Chem. 2019, 294, 9007–9015. [Google Scholar] [CrossRef]
- Pei, G.; Zyla, J.; He, L.; Moura-Alves, P.; Steinle, H.; Saikali, P.; Lozza, L.; Nieuwenhuizen, N.; Weiner, J.; Mollenkopf, H.-J.; et al. Cellular stress promotes NOD1/2-dependent inflammation via the endogenous metabolite sphingosine-1-phosphate. EMBO J. 2021, e106272. [Google Scholar] [CrossRef]
- Menu, P.; Mayor, A.; Zhou, R.; Tardivel, A.; Ichijo, H.; Mori, K.; Tschopp, J. ER stress activates the NLRP3 inflammasome via an UPR-independent pathway. Cell Death Dis. 2012, 3, e261. [Google Scholar] [CrossRef]
- Oslowski, C.M.; Hara, T.; O’Sullivan-Murphy, B.; Kanekura, K.; Lu, S.; Hara, M.; Ishigaki, S.; Zhu, L.J.; Hayashi, E.; Hui, S.T.; et al. Thioredoxin-interacting protein mediates ER stress-induced beta cell death through initiation of the inflammasome. Cell Metab. 2012, 16, 265–273. [Google Scholar] [CrossRef] [Green Version]
- Lerner, A.G.; Upton, J.P.; Praveen, P.V.; Ghosh, R.; Nakagawa, Y.; Igbaria, A.; Shen, S.; Nguyen, V.; Backes, B.J.; Heiman, M.; et al. IRE1alpha induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012, 16, 250–264. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Joe, Y.; Jeong, S.O.; Zheng, M.; Back, S.H.; Park, S.W.; Ryter, S.W.; Chung, H.T. Endoplasmic reticulum stress is sufficient for the induction of IL-1beta production via activation of the NF-kappaB and inflammasome pathways. Innate Immun. 2014, 20, 799–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tufanli, O.; Telkoparan Akillilar, P.; Acosta-Alvear, D.; Kocaturk, B.; Onat, U.I.; Hamid, S.M.; Cimen, I.; Walter, P.; Weber, C.; Erbay, E. Targeting IRE1 with small molecules counteracts progression of atherosclerosis. Proc. Natl. Acad. Sci. USA 2017, 114, E1395–E1404. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Dixon, B.J.; Doycheva, D.M.; Li, B.; Zhang, Y.; Hu, Q.; He, Y.; Guo, Z.; Nowrangi, D.; Flores, J.; et al. IRE1alpha inhibition decreased TXNIP/NLRP3 inflammasome activation through miR-17-5p after neonatal hypoxic-ischemic brain injury in rats. J. Neuroinflamm. 2018, 15, 32. [Google Scholar] [CrossRef] [PubMed]
- Bronner, D.N.; Abuaita, B.H.; Chen, X.; Fitzgerald, K.A.; Nunez, G.; He, Y.; Yin, X.M.; O’Riordan, M.X. Endoplasmic Reticulum Stress Activates the Inflammasome via NLRP3- and Caspase-2-Driven Mitochondrial Damage. Immunity 2015, 43, 451–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Z.; Pan, J.; Shen, Q.; Li, M.; Peng, Y. Mitochondrial dysfunction induces NLRP3 inflammasome activation during cerebral ischemia/reperfusion injury. J. Neuroinflamm. 2018, 15, 242. [Google Scholar] [CrossRef]
- Sarkar, S.; Malovic, E.; Harishchandra, D.S.; Ghaisas, S.; Panicker, N.; Charli, A.; Palanisamy, B.N.; Rokad, D.; Jin, H.; Anantharam, V.; et al. Mitochondrial impairment in microglia amplifies NLRP3 inflammasome proinflammatory signaling in cell culture and animal models of Parkinson’s disease. npj Parkinson’s Dis. 2017, 3, 30. [Google Scholar] [CrossRef] [PubMed]
- Gross, C.J.; Mishra, R.; Schneider, K.S.; Medard, G.; Wettmarshausen, J.; Dittlein, D.C.; Shi, H.; Gorka, O.; Koenig, P.A.; Fromm, S.; et al. K(+) Efflux-Independent NLRP3 Inflammasome Activation by Small Molecules Targeting Mitochondria. Immunity 2016, 45, 761–773. [Google Scholar] [CrossRef] [Green Version]
- Killackey, S.A.; Philpott, D.J.; Girardin, S.E. Mitophagy pathways in health and disease. J. Cell Biol. 2020, 219. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.J.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, L.D.; Pypaert, M.; Flavell, R.A.; Galan, J.E. A Salmonella protein causes macrophage cell death by inducing autophagy. J. Cell Biol. 2003, 163, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyden, E.D.; Dietrich, W.F. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat. Genet. 2006, 38, 240–244. [Google Scholar] [CrossRef]
- Chavarria-Smith, J.; Vance, R.E. Direct proteolytic cleavage of NLRP1B is necessary and sufficient for inflammasome activation by anthrax lethal factor. PLoS Pathog. 2013, 9, e1003452. [Google Scholar] [CrossRef] [Green Version]
- Chui, A.J.; Okondo, M.C.; Rao, S.D.; Gai, K.; Griswold, A.R.; Johnson, D.C.; Ball, D.P.; Taabazuing, C.Y.; Orth, E.L.; Vittimberga, B.A.; et al. N-terminal degradation activates the NLRP1B inflammasome. Science 2019, 364, 82–85. [Google Scholar] [CrossRef]
- Mitchell, P.S.; Sandstrom, A.; Vance, R.E. The NLRP1 inflammasome: New mechanistic insights and unresolved mysteries. Curr. Opin. Immunol. 2019, 60, 37–45. [Google Scholar] [CrossRef]
- Okondo, M.C.; Rao, S.D.; Taabazuing, C.Y.; Chui, A.J.; Poplawski, S.E.; Johnson, D.C.; Bachovchin, D.A. Inhibition of Dpp8/9 Activates the Nlrp1b Inflammasome. Cell Chem. Biol. 2018, 25, 262–267. [Google Scholar] [CrossRef]
- Zhong, F.L.; Robinson, K.; Teo, D.E.T.; Tan, K.Y.; Lim, C.; Harapas, C.R.; Yu, C.H.; Xie, W.H.; Sobota, R.M.; Au, V.B.; et al. Human DPP9 represses NLRP1 inflammasome and protects against autoinflammatory diseases via both peptidase activity and FIIND domain binding. J. Biol. Chem. 2018, 293, 18864–18878. [Google Scholar] [CrossRef] [Green Version]
- Taabazuing, C.Y.; Griswold, A.R.; Bachovchin, D.A. The NLRP1 and CARD8 inflammasomes. Immunol. Rev. 2020, 297, 13–25. [Google Scholar] [CrossRef]
- Liao, K.C.; Mogridge, J. Activation of the Nlrp1b inflammasome by reduction of cytosolic ATP. Infect. Immun. 2013, 81, 570–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matzinger, P. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 1994, 12, 991–1045. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Tsuda, K.; Parker, J.E. Effector-triggered immunity: From pathogen perception to robust defense. Annu. Rev. Plant Biol. 2015, 66, 487–511. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.D.; Vance, R.E.; Dangl, J.L. Intracellular innate immune surveillance devices in plants and animals. Science 2016, 354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Pathogens cause cytoskeleton disruption. Bacteria and viruses subvert host cytoskeleton dynamics for their entry, intracellular survival and dissemination. They employ various effector molecules, toxins, or viral proteins to induce actin remodeling by manipulating Rho GTPases (RHOA, CDC42, and RAC1), which are the central regulators of the actin polymerization. Parasites such as Toxoplasma gondii and Plasmodium berghei also induce actin rearrangement by binding or cleaving actin-associated factors. Abbreviations: RHOA, Ras homolog family member A; CDC42, cell division control protein 42 homolog; RAC1, Ras-related C3 botulinum toxin substrate 1; mDia, mammalian homolog of Diaphanous; N-WASP, neuronal Wiskott-Aldrich Syndrome protein; WAVE, Wiskott-Aldrich syndrome protein family verprolin-homologous; ARP2/3, actin related protein 2/3; TirA, translocated intimin receptor A; EspF, enteropathogenic E. coli effector protein F; EspG, enteropathogenic E. coli effector protein G; TcdA, Clostridium difficile toxin A; TcdB, C. difficile toxin B; IpaC, invasion plasmid antigen C; VirA, virulence factor A; SopE, salmonella outer protein E; SipA, salmonella invasion protein A; TARP, type III secretion system actin-recruiting effector. Image created with BioRender.com.
Figure 1.
Pathogens cause cytoskeleton disruption. Bacteria and viruses subvert host cytoskeleton dynamics for their entry, intracellular survival and dissemination. They employ various effector molecules, toxins, or viral proteins to induce actin remodeling by manipulating Rho GTPases (RHOA, CDC42, and RAC1), which are the central regulators of the actin polymerization. Parasites such as Toxoplasma gondii and Plasmodium berghei also induce actin rearrangement by binding or cleaving actin-associated factors. Abbreviations: RHOA, Ras homolog family member A; CDC42, cell division control protein 42 homolog; RAC1, Ras-related C3 botulinum toxin substrate 1; mDia, mammalian homolog of Diaphanous; N-WASP, neuronal Wiskott-Aldrich Syndrome protein; WAVE, Wiskott-Aldrich syndrome protein family verprolin-homologous; ARP2/3, actin related protein 2/3; TirA, translocated intimin receptor A; EspF, enteropathogenic E. coli effector protein F; EspG, enteropathogenic E. coli effector protein G; TcdA, Clostridium difficile toxin A; TcdB, C. difficile toxin B; IpaC, invasion plasmid antigen C; VirA, virulence factor A; SopE, salmonella outer protein E; SipA, salmonella invasion protein A; TARP, type III secretion system actin-recruiting effector. Image created with BioRender.com.

Figure 2.
Various pathogens cause ER stress to modulate host cell death or facilitate their replication and dissemination. Unfolded protein responses (UPR) are mediated by the ER stress sensors protein kinase R-like ER kinase (PERK), inositol-requiring enzyme 1α (IRE1α) and activating transcription factor 6 (ATF6). The 6 kDa early secretory antigenic target (ESAT-6) from M. tuberculosis (Mtb), shiga toxin 1 (Stx) from S. dysenteriae, listeriolysin O (LLO) from L. monocytogenes, dengue virus (DENV), etc. induce all three branches of UPR. VceC, a T4SS effector from B. abortus, binds to the ER chaperon binding immunoglobulin protein (BiP) and triggers IRE1α signaling. CaeB (C. burnetii anti-apoptotic effector B) from C. burnetii stimulates IRE1α signaling, facilitating its pathogenesis. The severe acute respiratory syndrome coronavirus (SARS-CoV) induces PERK and ATF6 signaling and the African Swine Fever Virus (ASFV) activates only ATF6. Image created with BioRender.com.
Figure 2.
Various pathogens cause ER stress to modulate host cell death or facilitate their replication and dissemination. Unfolded protein responses (UPR) are mediated by the ER stress sensors protein kinase R-like ER kinase (PERK), inositol-requiring enzyme 1α (IRE1α) and activating transcription factor 6 (ATF6). The 6 kDa early secretory antigenic target (ESAT-6) from M. tuberculosis (Mtb), shiga toxin 1 (Stx) from S. dysenteriae, listeriolysin O (LLO) from L. monocytogenes, dengue virus (DENV), etc. induce all three branches of UPR. VceC, a T4SS effector from B. abortus, binds to the ER chaperon binding immunoglobulin protein (BiP) and triggers IRE1α signaling. CaeB (C. burnetii anti-apoptotic effector B) from C. burnetii stimulates IRE1α signaling, facilitating its pathogenesis. The severe acute respiratory syndrome coronavirus (SARS-CoV) induces PERK and ATF6 signaling and the African Swine Fever Virus (ASFV) activates only ATF6. Image created with BioRender.com.

Figure 3.
Mitochondrial dysfunction and blockade of host translation induced by pathogens. (A) Manipulation of mitochondrial dynamics by pathogens. Pathogenic viruses and bacteria cause mitochondrial fragmentation or mitochondrial fusion for replication, persistence in host cell or dissemination; (B) Modulation of host metabolism by pathogens. To acquire the nutrient from the host, pathogens trigger glycolysis or modulate the TCA cycle dependent on their metabolic requirements; (C) Inhibition of host translation by pathogens. To shut off host defense, many viruses such as poliovirus, coxsackievirus and rhinovirus, HIV-1, enteroviruses, and SARS-CoV2 target the eIF4F complex to inhibit translation initiation, while some bacterial pathogens utilize effector proteins or toxins to inactivate translation elongation factors-EF1A and EF2. Abbreviations: DRP1, dynamin related protein 1; VacA, vacuolating cytotoxin A; LLO, listeriolysin O; TLR2, toll-like receptor 2; AKT, protein kinase B; mTOR, mechanistic target of Rapamycin; TCA cycle, tricarboxylic acid cycle; HCMV, Human Cytomegalovirus; HCV, Hepatitis C Virus; HSV-1, Herpes simplex virus 1; HIV, Human immunodeficiency virus; MMLV, Moloney murine leukemia virus; MMTV, Mouse mammary tumor virus; SIV, Simian immunodeficiency virus; FMDV, Foot-and-mouth disease virus; eIF, eukaryotic translation initiation factor; EF1A/1B, eukaryotic translation elongation factor 1A/1B; Lgt1, Legionella glucosyltransferase 1; SidI, substrate I of Icm/Dot transporter; Nsp1, nonstructural protein 1. Image created with BioRender.com.
Figure 3.
Mitochondrial dysfunction and blockade of host translation induced by pathogens. (A) Manipulation of mitochondrial dynamics by pathogens. Pathogenic viruses and bacteria cause mitochondrial fragmentation or mitochondrial fusion for replication, persistence in host cell or dissemination; (B) Modulation of host metabolism by pathogens. To acquire the nutrient from the host, pathogens trigger glycolysis or modulate the TCA cycle dependent on their metabolic requirements; (C) Inhibition of host translation by pathogens. To shut off host defense, many viruses such as poliovirus, coxsackievirus and rhinovirus, HIV-1, enteroviruses, and SARS-CoV2 target the eIF4F complex to inhibit translation initiation, while some bacterial pathogens utilize effector proteins or toxins to inactivate translation elongation factors-EF1A and EF2. Abbreviations: DRP1, dynamin related protein 1; VacA, vacuolating cytotoxin A; LLO, listeriolysin O; TLR2, toll-like receptor 2; AKT, protein kinase B; mTOR, mechanistic target of Rapamycin; TCA cycle, tricarboxylic acid cycle; HCMV, Human Cytomegalovirus; HCV, Hepatitis C Virus; HSV-1, Herpes simplex virus 1; HIV, Human immunodeficiency virus; MMLV, Moloney murine leukemia virus; MMTV, Mouse mammary tumor virus; SIV, Simian immunodeficiency virus; FMDV, Foot-and-mouth disease virus; eIF, eukaryotic translation initiation factor; EF1A/1B, eukaryotic translation elongation factor 1A/1B; Lgt1, Legionella glucosyltransferase 1; SidI, substrate I of Icm/Dot transporter; Nsp1, nonstructural protein 1. Image created with BioRender.com.

Figure 4.
NOD1 and NOD2 trigger NF-κB activation by detecting pathogen-induced cytoskeleton disruption or pathogen-induced ER stress. Virulence factors from S. typhimurium and S. flexneri induce aberrant actin polymerization, subsequently triggering NOD1-mediated NF-κB activation. B. abortus and C. muridarum elicit ER stress, which is further sensed by NOD1 and NOD2 to enable NF-κB activation. Abbreviations: RHOA, Ras homolog family member A; CDC42, cell division control protein 42 homolog; RAC1, Ras-related C3 botulinum toxin substrate 1; GEF-H1, guanine nucleotide exchange factor H1; TRAF2, TNF receptor associated factor 2; RIP2, receptor-interacting-serine/threonine-protein kinase 2; IKB, inhibitor of κB; IRE1α, inositol-requiring enzyme-1a; IpgB2, invasion plasmid gene B2; SopE, Salmonella outer protein E; VceC, virB-coregulated effector C. Image created with BioRender.com.
Figure 4.
NOD1 and NOD2 trigger NF-κB activation by detecting pathogen-induced cytoskeleton disruption or pathogen-induced ER stress. Virulence factors from S. typhimurium and S. flexneri induce aberrant actin polymerization, subsequently triggering NOD1-mediated NF-κB activation. B. abortus and C. muridarum elicit ER stress, which is further sensed by NOD1 and NOD2 to enable NF-κB activation. Abbreviations: RHOA, Ras homolog family member A; CDC42, cell division control protein 42 homolog; RAC1, Ras-related C3 botulinum toxin substrate 1; GEF-H1, guanine nucleotide exchange factor H1; TRAF2, TNF receptor associated factor 2; RIP2, receptor-interacting-serine/threonine-protein kinase 2; IKB, inhibitor of κB; IRE1α, inositol-requiring enzyme-1a; IpgB2, invasion plasmid gene B2; SopE, Salmonella outer protein E; VceC, virB-coregulated effector C. Image created with BioRender.com.

Figure 5.
Distinct NLRP inflammasomes are activated by ER stress, mitochondrial dysfunction or directly by pathogens. (A) B. abortus induces ER stress, which causes NLRP3 inflammasome activation. (B) S. typhimurium targets mitochondria to trigger NLRP3 inflammasome activation. (C) Murine and human NLRP1 (mNLRP1B, hNLRP1) are targeted by pathogen virulence factors. Abbreviations: PERK, PKR-like ER kinase; IRE1α, inositol-requiring enzyme 1α; ATF6, activating transcription factor 6; TXNIP, thioredoxin-interacting protein; ASC, apoptosis-associated Speck-like protein containing a CARD; GSDMD, Gasdermin D; UBR2, ubiquitin protein ligase E3 component N-Recognin 2; ROS, reactive oxygen species; mtDNA, mitochondrial DNA; SipB, Salmonella invasion protein B; IpaH7.8, invasion plasmid antigen H7.8. Image created with BioRender.com.
Figure 5.
Distinct NLRP inflammasomes are activated by ER stress, mitochondrial dysfunction or directly by pathogens. (A) B. abortus induces ER stress, which causes NLRP3 inflammasome activation. (B) S. typhimurium targets mitochondria to trigger NLRP3 inflammasome activation. (C) Murine and human NLRP1 (mNLRP1B, hNLRP1) are targeted by pathogen virulence factors. Abbreviations: PERK, PKR-like ER kinase; IRE1α, inositol-requiring enzyme 1α; ATF6, activating transcription factor 6; TXNIP, thioredoxin-interacting protein; ASC, apoptosis-associated Speck-like protein containing a CARD; GSDMD, Gasdermin D; UBR2, ubiquitin protein ligase E3 component N-Recognin 2; ROS, reactive oxygen species; mtDNA, mitochondrial DNA; SipB, Salmonella invasion protein B; IpaH7.8, invasion plasmid antigen H7.8. Image created with BioRender.com.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pei, G.; Dorhoi, A. NOD-Like Receptors: Guards of Cellular Homeostasis Perturbation during Infection. Int. J. Mol. Sci. 2021, 22, 6714. https://doi.org/10.3390/ijms22136714
AMA Style
Pei G, Dorhoi A. NOD-Like Receptors: Guards of Cellular Homeostasis Perturbation during Infection. International Journal of Molecular Sciences. 2021; 22(13):6714. https://doi.org/10.3390/ijms22136714
Chicago/Turabian StylePei, Gang, and Anca Dorhoi. 2021. "NOD-Like Receptors: Guards of Cellular Homeostasis Perturbation during Infection" International Journal of Molecular Sciences 22, no. 13: 6714. https://doi.org/10.3390/ijms22136714
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.