Evolution of Urothelial Bladder Cancer in the Context of Molecular Classifications

 and
and
Abstract
:1. Introduction: The Clinical Problem of Bladder Cancer
2. Current BLCa Management
2.1. NMIBC Treatment Options and Follow-Up
2.2. MIBC Treatment Options and Follow-Up
3. Lack of Predictive Biomarkers: First Cause of Treatment Failure
4. Molecular Classification
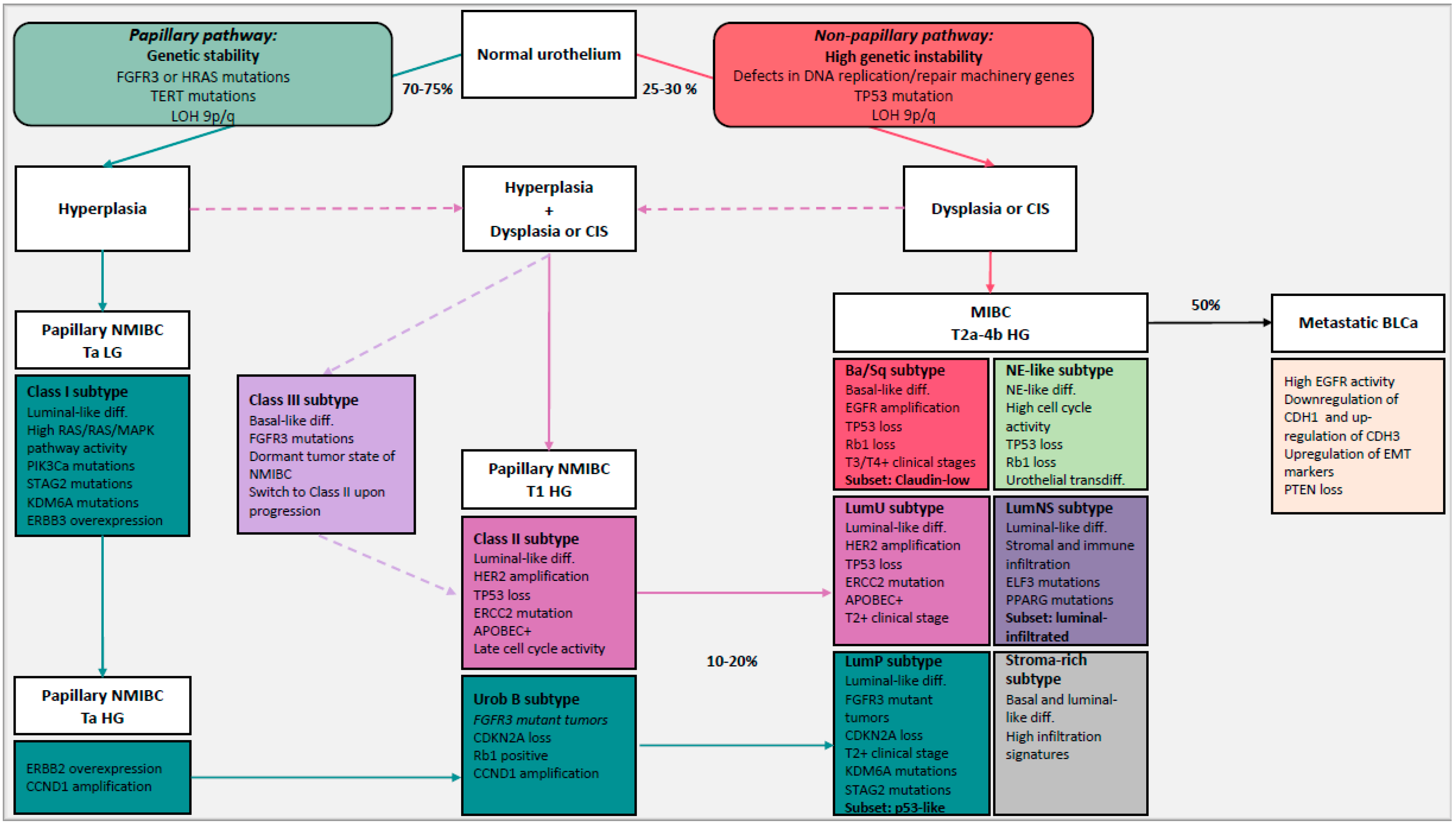
4.1. NMIBC and MIBC: Two-Pathway Theory
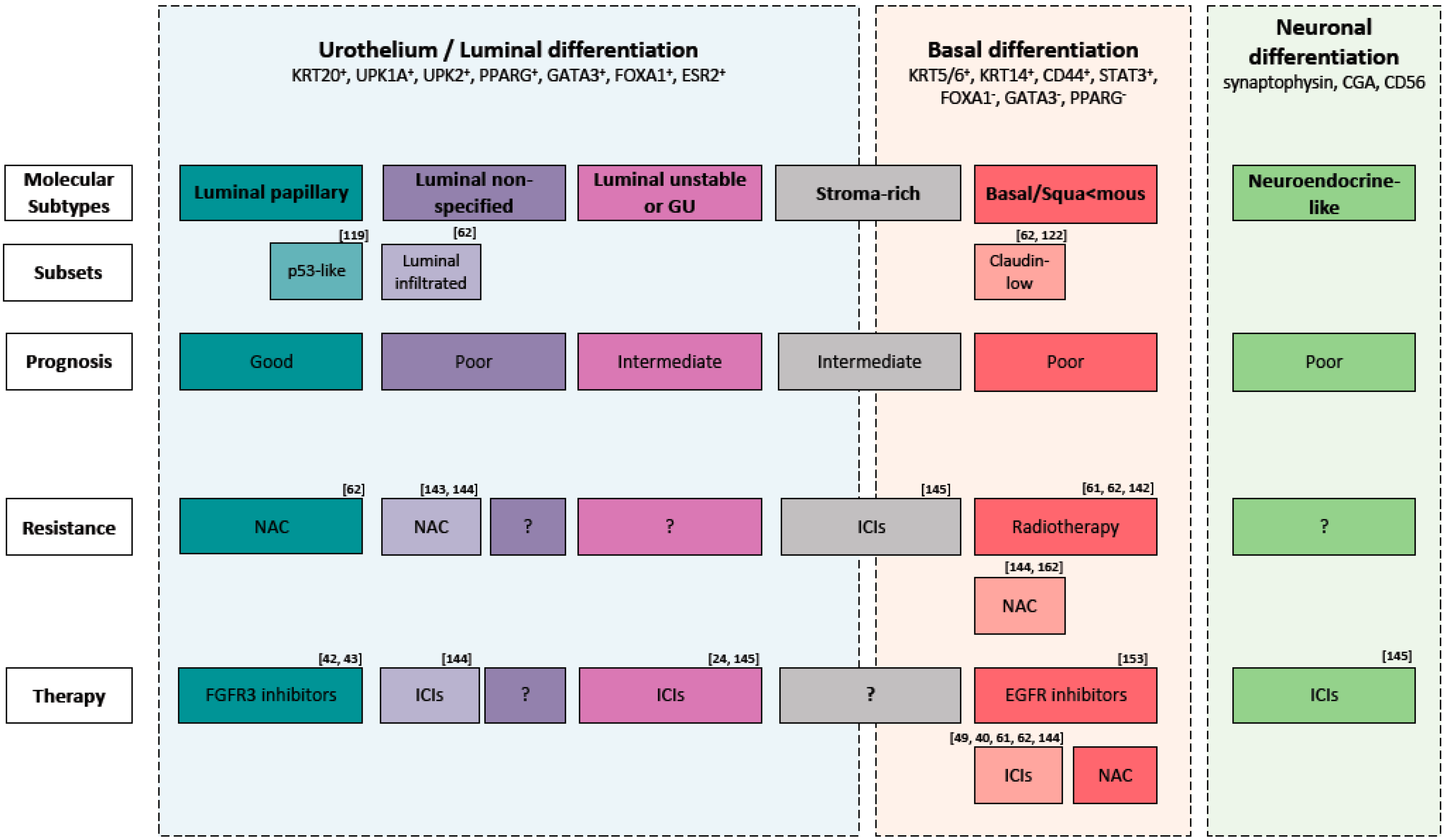
4.2. Molecular Subtypes to Understand BLCa Biology
5. Molecular Classification to Guide Treatment Choice
6. Limitations and Perspectives of Molecular Classification
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, B.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Letašiová, S.; Medveďová, A.; Šovčíková, A.; Dušinská, M.; Volkovová, K.; Mosoiu, C.; Bartonová, A. Bladder Cancer, a Review of the Environmental Risk Factors. Environ. Health 2012, 11 (Suppl. 1), S11. [Google Scholar]
- Burger, M.; Catto, J.W.F.; Dalbagni, G.; Grossman, H.B.; Herr, H.; Karakiewicz, P.; Kassouf, W.; Kiemeney, L.A.; la Vecchia, C.; Shariat, S.; et al. Epidemiology and Risk Factors of Urothelial Bladder Cancer. Eur. Urol. 2013, 63, 234–241. [Google Scholar] [CrossRef]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Dyba, T.; Randi, G.; Bettio, M.; Gavin, A.; Visser, O.; Bray, F. Cancer Incidence and Mortality Patterns in Europe: Estimates for 40 Countries and 25 Major Cancers in 2018. Eur. J. Cancer 2018, 103, 356–387. [Google Scholar] [CrossRef] [PubMed]
- Berdik, C. Unlocking Bladder Cancer. Nature 2017, 551, S34–S35. [Google Scholar] [CrossRef] [PubMed]
- Aben, K.K.H.; Witjes, J.A.; Schoenberg, M.P.; Kaa, C.H.d.; Verbeek, A.L.M.; Kiemeney, L.A.L.M. Familial Aggregation of Urothelial Cell Carcinoma. Int. J. Cancer 2002, 98, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Spitz, M.R.; Dinney, C.P.; Etzel, C.J.; Grossman, H.B.; Wu, X. Bladder Cancer Risk as Modified by Family History and Smoking. Cancer 2006, 107, 705–711. [Google Scholar] [CrossRef]
- Murta-Nascimento, C.; Silverman, D.T.; Kogevinas, M.; García-Closas, M.; Rothman, N.; Tardón, A.; García-Closas, R.; Serra, C.; Carrato, A.; Villanueva, C.; et al. Risk of Bladder Cancer Associated with Family History of Cancer: Do Low-Penetrance Polymorphisms Account for the Increase in Risk? Cancer Epidemiol. Prev. Biomark. 2007, 16, 1595–1600. [Google Scholar] [CrossRef] [Green Version]
- Svatek, R.S.; Hollenbeck, B.K.; Holmäng, S.; Lee, R.; Kim, S.P.; Stenzl, A.; Lotan, Y. The Economics of Bladder Cancer: Costs and Considerations of Caring for This Disease. Eur. Urol. 2014, 66, 253–262. [Google Scholar] [CrossRef]
- Hong, Y.M.; Loughlin, K.R. Economic Impact of Tumor Markers in Bladder Cancer Surveillance. Urology 2008, 71, 131–135. [Google Scholar] [CrossRef]
- James, A.C.; Gore, J.L. The Costs of Non-Muscle Invasive Bladder Cancer. Urol. Clin. N. Am. 2013, 40, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Ploeg, M.; Aben, K.K.H.; Kiemeney, L.A. The Present and Future Burden of Urinary Bladder Cancer in the World. World J. Urol. 2009, 27, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avritscher, E.B.C.; Cooksley, C.D.; Grossman, H.B.; Sabichi, A.L.; Hamblin, L.; Dinney, C.P.; Elting, L.S. Clinical Model of Lifetime Cost of Treating Bladder Cancer and Associated Complications. Urology 2006, 68, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Kamat, A.M.; Hahn, N.M.; Efstathiou, J.A.; Lerner, S.P.; Malmström, P.-U.; Choi, W.; Guo, C.C.; Lotan, Y.; Kassouf, W. Bladder Cancer. Lancet 2016, 388, 2796–2810. [Google Scholar] [CrossRef]
- Rhijn, B.W.G.v.; Burger, M.; Lotan, Y.; Solsona, E.; Stief, C.G.; Sylvester, R.J.; Witjes, J.A.; Zlotta, A.R. Recurrence and Progression of Disease in Non–Muscle-Invasive Bladder Cancer: From Epidemiology to Treatment Strategy. Eur. Urol. 2009, 56, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Chamie, K.; Litwin, M.S.; Bassett, J.C.; Daskivich, T.J.; Lai, J.; Hanley, J.M.; Konety, B.R.; Saigal, C.S.; Urologic Diseases in America Project. Recurrence of High-Risk Bladder Cancer: A Population-Based Analysis. Cancer 2013, 119, 3219–3227. [Google Scholar] [CrossRef] [Green Version]
- Sylvester, R.J.; van der Meijden, A.P.M.; Oosterlinck, W.; Witjes, J.A.; Bouffioux, C.; Denis, L.; Newling, D.W.W.; Kurth, K. Predicting Recurrence and Progression in Individual Patients with Stage Ta T1 Bladder Cancer Using EORTC Risk Tables: A Combined Analysis of 2596 Patients from Seven EORTC Trials. Eur. Urol. 2006, 49, 466–477. [Google Scholar] [CrossRef]
- van den Bosch, S.; Alfred Witjes, J. Long-Term Cancer-Specific Survival in Patients with High-Risk, Non-Muscle-Invasive Bladder Cancer and Tumour Progression: A Systematic Review. Eur. Urol. 2011, 60, 493–500. [Google Scholar] [CrossRef]
- van Rhijn, B.W.G.; van der Kwast, T.H.; Alkhateeb, S.S.; Fleshner, N.E.; van Leenders, G.J.L.H.; Bostrom, P.J.; van der Aa, M.N.M.; Kakiashvili, D.M.; Bangma, C.H.; Jewett, M.A.S.; et al. New and Highly Prognostic System to Discern T1 Bladder Cancer Substage. Eur. Urol. 2012, 61, 378–384. [Google Scholar] [CrossRef]
- Tang, D.H.; Chang, S.S. Management of Carcinoma in Situ of the Bladder: Best Practice and Recent Developments. Ther. Adv. Urol. 2015, 7, 351–364. [Google Scholar] [CrossRef] [Green Version]
- Knowles, M.A.; Hurst, C.D. Molecular Biology of Bladder Cancer: New Insights into Pathogenesis and Clinical Diversity. Nat. Rev. Cancer 2015, 15, 25–41. [Google Scholar] [CrossRef] [PubMed]
- Shinagare, A.B.; Ramaiya, N.H.; Jagannathan, J.P.; Fennessy, F.M.; Taplin, M.; van den Abbeele, A.D. Metastatic Pattern of Bladder Cancer: Correlation With the Characteristics of the Primary Tumor. Am. J. Roentgenol. 2011, 196, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Hautmann, R.E.; Gschwend, J.E.; de Petriconi, R.C.; Kron, M.; Volkmer, B.G. Cystectomy for Transitional Cell Carcinoma of the Bladder: Results of a Surgery Only Series in the Neobladder Era. J. Urol. 2006, 176, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.J.; Cho, K.S.; Koo, K.C. Current Status and Future Perspectives of Immunotherapy for Locally Advanced or Metastatic Urothelial Carcinoma: A Comprehensive Review. Cancers 2020, 12, 192. [Google Scholar] [CrossRef] [Green Version]
- Babjuk, M.; Burger, M.; Compérat, E.M.; Gontero, P.; Mostafid, A.H.; Palou, J.; van Rhijn, B.W.G.; Rouprêt, M.; Shariat, S.F.; Sylvester, R.; et al. European Association of Urology Guidelines on Non-Muscle-Invasive Bladder Cancer (TaT1 and Carcinoma In Situ)—2019 Update. Eur. Urol. 2019, 76, 639–657. [Google Scholar] [CrossRef]
- Soukup, V.; Čapoun, O.; Cohen, D.; Hernández, V.; Babjuk, M.; Burger, M.; Compérat, E.; Gontero, P.; Lam, T.; MacLennan, S.; et al. Prognostic Performance and Reproducibility of the 1973 and 2004/2016 World Health Organization Grading Classification Systems in Non–Muscle-Invasive Bladder Cancer: A European Association of Urology Non-Muscle Invasive Bladder Cancer Guidelines Panel Systematic Review. Eur. Urol. 2017, 72, 801–813. [Google Scholar]
- Lipunova, N.; Wesselius, A.; Cheng, K.K.; van Schooten, F.J.; Cazier, J.-B.; Bryan, R.T.; Zeegers, M.P. Systematic Review: Genetic Associations for Prognostic Factors of Urinary Bladder Cancer. Biomark. Cancer 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Babjuk, M.; Böhle, A.; Burger, M.; Capoun, O.; Cohen, D.; Compérat, E.M.; Hernández, V.; Kaasinen, E.; Palou, J.; Rouprêt, M.; et al. EAU Guidelines on Non–Muscle-Invasive Urothelial Carcinoma of the Bladder: Update 2016. Eur. Urol. 2017, 71, 447–461. [Google Scholar] [CrossRef]
- Clark, P.E.; Agarwal, N.; Biagioli, M.C.; Eisenberger, M.A.; Greenberg, R.E.; Herr, H.W.; Inman, B.A.; Kuban, D.A.; Kuzel, T.M.; Lele, S.M.; et al. Bladder Cancer. J. Natl. Compr. Canc. Netw. 2013, 11, 446–475. [Google Scholar] [CrossRef] [Green Version]
- Holmäng, S.; Ströck, V. Should Follow-up Cystoscopy in Bacillus Calmette-Guérin–Treated Patients Continue After Five Tumour-Free Years? Eur. Urol. 2012, 61, 503–507. [Google Scholar] [CrossRef]
- Perlis, N.; Zlotta, A.R.; Beyene, J.; Finelli, A.; Fleshner, N.E.; Kulkarni, G.S. Immediate Post–Transurethral Resection of Bladder Tumor Intravesical Chemotherapy Prevents Non–Muscle-Invasive Bladder Cancer Recurrences: An Updated Meta-Analysis on 2548 Patients and Quality-of-Evidence Review. Eur. Urol. 2013, 64, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Arends, T.J.H.; Nativ, O.; Maffezzini, M.; de Cobelli, O.; Canepa, G.; Verweij, F.; Moskovitz, B.; van der Heijden, A.G.; Witjes, J.A. Results of a Randomised Controlled Trial Comparing Intravesical Chemohyperthermia with Mitomycin C Versus Bacillus Calmette-Guérin for Adjuvant Treatment of Patients with Intermediate- and High-Risk Non–Muscle-Invasive Bladder Cancer. Eur. Urol. 2016, 69, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Kamat, A.M.; Flaig, T.W.; Grossman, H.B.; Konety, B.; Lamm, D.; O’Donnell, M.A.; Uchio, E.; Efstathiou, J.A.; Taylor, J.A. Consensus Statement on Best Practice Management Regarding the Use of Intravesical Immunotherapy with BCG for Bladder Cancer. Nat. Rev. Urol. 2015, 12, 225–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sylvester, R.J.; Oosterlinck, W.; Holmang, S.; Sydes, M.R.; Birtle, A.; Gudjonsson, S.; de Nunzio, C.; Okamura, K.; Kaasinen, E.; Solsona, E.; et al. Systematic Review and Individual Patient Data Meta-Analysis of Randomized Trials Comparing a Single Immediate Instillation of Chemotherapy After Transurethral Resection with Transurethral Resection Alone in Patients with Stage PTa–PT1 Urothelial Carcinoma of the Bladder: Which Patients Benefit from the Instillation? Eur. Urol. 2016, 69, 231–244. [Google Scholar]
- Sylvester, R.J.; van der Meijden, A.P.M.; Lamm, D.L. Intravesical Bacillus Calmette-Guerin Reduces the Risk of Progression in Patients with Superficial Bladder Cancer: A Meta-Analysis of the Published Results of Randomized Clinical Trials. J. Urol. 2002, 168, 1964–1970. [Google Scholar] [CrossRef]
- Witjes, J.A.; Bruins, H.M.; Cathomas, R.; Compérat, E.M.; Cowan, N.C.; Gakis, G.; Hernández, V.; Linares Espinós, E.; Lorch, A.; Neuzillet, Y.; et al. European Association of Urology Guidelines on Muscle-Invasive and Metastatic Bladder Cancer: Summary of the 2020 Guidelines. Eur. Urol. 2020. [Google Scholar] [CrossRef]
- Sonpavde, G.; Watson, D.; Tourtellott, M.; Cowey, C.L.; Hellerstedt, B.; Hutson, T.E.; Zhan, F.; Vogelzang, N.J. Administration of Cisplatin-Based Chemotherapy for Advanced Urothelial Carcinoma in the Community. Clin. Genitourin. Cancer 2012, 10, 1–5. [Google Scholar] [CrossRef]
- Zargar, H.; Espiritu, P.N.; Fairey, A.S.; Mertens, L.S.; Dinney, C.P.; Mir, M.C.; Krabbe, L.-M.; Cookson, M.S.; Jacobsen, N.-E.; Gandhi, N.; et al. Multicenter Assessment of Neoadjuvant Chemotherapy for Muscle-Invasive Bladder Cancer. Eur. Urol. 2015, 67, 241–249. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; van der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in Patients with Locally Advanced and Metastatic Urothelial Carcinoma Who Have Progressed Following Treatment with Platinum-Based Chemotherapy: A Single-Arm, Multicentre, Phase 2 Trial. Lancet Lond. Engl. 2016, 387, 1909–1920. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Callahan, M.K.; Bono, P.; Kim, J.; Spiliopoulou, P.; Calvo, E.; Pillai, R.N.; Ott, P.A.; de Braud, F.; Morse, M.; et al. Nivolumab Monotherapy in Recurrent Metastatic Urothelial Carcinoma (CheckMate 032): A Multicentre, Open-Label, Two-Stage, Multi-Arm, Phase 1/2 Trial. Lancet Oncol. 2016, 17, 1590–1598. [Google Scholar] [CrossRef] [Green Version]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune Checkpoint Blockade Therapy for Cancer: An Overview of FDA-Approved Immune Checkpoint Inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Montazeri, K.; Bellmunt, J. Erdafitinib for the Treatment of Metastatic Bladder Cancer. Expert Rev. Clin. Pharmacol. 2020, 13, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Loriot, Y.; Necchi, A.; Park, S.H.; Garcia-Donas, J.; Huddart, R.; Burgess, E.; Fleming, M.; Rezazadeh, A.; Mellado, B.; Varlamov, S.; et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2019, 381, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.E.; O’Donnell, P.H.; Balar, A.V.; McGregor, B.A.; Heath, E.I.; Yu, E.Y.; Galsky, M.D.; Hahn, N.M.; Gartner, E.M.; Pinelli, J.M.; et al. Pivotal Trial of Enfortumab Vedotin in Urothelial Carcinoma After Platinum and Anti-Programmed Death 1/Programmed Death Ligand 1 Therapy. J. Clin. Oncol. 2019, 37, 2592–2600. [Google Scholar] [CrossRef]
- Bosschieter, J.; Nieuwenhuijzen, J.A.; van Ginkel, T.; Vis, A.N.; Witte, B.; Newling, D.; Beckers, G.M.A.; van Moorselaar, R.J.A. Value of an Immediate Intravesical Instillation of Mitomycin C in Patients with Non–Muscle-Invasive Bladder Cancer: A Prospective Multicentre Randomised Study in 2243 Patients. Eur. Urol. 2018, 73, 226–232. [Google Scholar] [CrossRef]
- Steinberg, R.L.; Thomas, L.J.; O’Donnell, M.A. Bacillus Calmette-Guérin (BCG) Treatment Failures in Non-Muscle Invasive Bladder Cancer: What Truly Constitutes Unresponsive Disease. Bladder Cancer 2015, 1, 105–116. [Google Scholar] [CrossRef] [Green Version]
- Lamm, D.; Persad, R.; Brausi, M.; Buckley, R.; Witjes, J.A.; Palou, J.; Böhle, A.; Kamat, A.M.; Colombel, M.; Soloway, M. Defining Progression in Nonmuscle Invasive Bladder Cancer: It Is Time for a New, Standard Definition. J. Urol. 2014, 191, 20–27. [Google Scholar] [CrossRef]
- Power, C.A.; Wei, G.; Bretscher, P.A. Mycobacterial Dose Defines the Th1/Th2 Nature of the Immune Response Independently of Whether Immunization Is Administered by the Intravenous, Subcutaneous, or Intradermal Route. Infect. Immun. 1998, 66, 5743–5750. [Google Scholar] [CrossRef] [Green Version]
- Zlotta, A.R.; Fleshner, N.E.; Jewett, M.A. The Management of BCG Failure in Non-Muscle-Invasive Bladder Cancer: An Update. Can. Urol. Assoc. J. 2009, 3 (Suppl. 4), S199–S205. [Google Scholar] [CrossRef]
- Kates, M.; Matoso, A.; Choi, W.; Baras, A.S.; Daniels, M.J.; Lombardo, K.; Brant, A.; Mikkilineni, N.; McConkey, D.J.; Kamat, A.M.; et al. Adaptive Immune Resistance to Intravesical BCG in Non-Muscle Invasive Bladder Cancer: Implications for Prospective BCG Unresponsive Trials. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Pembrolizumab and BCG Solution in Treating Patients With Recurrent Non-Muscle-Invasive Bladder Cancer—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02808143 (accessed on 10 July 2020).
- Ślusarczyk, A.; Zapała, P.; Zapała, Ł.; Piecha, T.; Radziszewski, P. Prediction of BCG Responses in Non-Muscle-Invasive Bladder Cancer in the Era of Novel Immunotherapeutics. Int. Urol. Nephrol. 2019, 51, 1089–1099. [Google Scholar] [CrossRef] [PubMed]
- International Collaboration of Trialists on Behalf of the Medical Research Council Advanced Bladder Cancer Working Party; EORTC Genito-Urinary Group; Australian Bladder Cancer Study Group; National Cancer Institute of Canada Clinical Trials Group; Finnbladder, Norwegian Bladder Cancer Study Group; Club Urologico Espanol de Tratamiento Oncologico (CUETO) Group. Neoadjuvant Cisplatin, Methotrexate, and Vinblastine Chemotherapy for Muscle-Invasive Bladder Cancer: A Randomised Controlled Trial. Lancet 1999, 354, 533–540. [Google Scholar] [CrossRef]
- International Collaboration of Trialists. International Phase III Trial Assessing Neoadjuvant Cisplatin, Methotrexate, and Vinblastine Chemotherapy for Muscle-Invasive Bladder Cancer: Long-Term Results of the BA06 30894 Trial. J. Clin. Oncol. 2011, 29, 2171–2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plimack, E.R.; Hoffman-Censits, J.H.; Viterbo, R.; Trabulsi, E.J.; Ross, E.A.; Greenberg, R.E.; Chen DY, T.; Lallas, C.D.; Wong, Y.-N.; Lin, J.; et al. Accelerated Methotrexate, Vinblastine, Doxorubicin, and Cisplatin Is Safe, Effective, and Efficient Neoadjuvant Treatment for Muscle-Invasive Bladder Cancer: Results of a Multicenter Phase II Study With Molecular Correlates of Response and Toxicity. J. Clin. Oncol. 2014, 32, 1895–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choueiri, T.K.; Jacobus, S.; Bellmunt, J.; Qu, A.; Appleman, L.J.; Tretter, C.; Bubley, G.J.; Stack, E.C.; Signoretti, S.; Walsh, M.; et al. Neoadjuvant Dose-Dense Methotrexate, Vinblastine, Doxorubicin, and Cisplatin With Pegfilgrastim Support in Muscle-Invasive Urothelial Cancer: Pathologic, Radiologic, and Biomarker Correlates. J. Clin. Oncol. 2014, 32, 1889–1894. [Google Scholar] [CrossRef]
- Shah, J.B.; McConkey, D.J.; Dinney, C.P.N. New Strategies in Muscle-Invasive Bladder Cancer: On the Road to Personalized Medicine. Clin. Cancer Res. 2011, 17, 2608–2612. [Google Scholar] [CrossRef] [Green Version]
- Fahmy, N.M.; Mahmud, S.; Aprikian, A.G. Delay in the Surgical Treatment of Bladder Cancer and Survival: Systematic Review of the Literature. Eur. Urol. 2006, 50, 1176–1182. [Google Scholar] [CrossRef]
- Kassouf, W.; Agarwal, P.K.; Grossman, H.B.; Leibovici, D.; Munsell, M.F.; Siefker-Radtke, A.; Pisters, L.L.; Swanson, D.A.; Dinney, C.P.N.; Kamat, A.M. Outcome of Patients with Bladder Cancer with PN+ Disease after Preoperative Chemotherapy and Radical Cystectomy. Urology 2009, 73, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Dalbagni, G.; Vora, K.; Kaag, M.; Cronin, A.; Bochner, B.; Donat, S.M.; Herr, H.W. Clinical Outcome in a Contemporary Series of Restaged Patients with Clinical T1 Bladder Cancer. Eur. Urol. 2009, 56, 903–910. [Google Scholar] [CrossRef]
- McConkey, D.J.; Choi, W.; Shen, Y.; Lee, I.-L.; Porten, S.; Matin, S.F.; Kamat, A.M.; Corn, P.; Millikan, R.E.; Dinney, C.; et al. A Prognostic Gene Expression Signature in the Molecular Classification of Chemotherapy-Naïve Urothelial Cancer Is Predictive of Clinical Outcomes from Neoadjuvant Chemotherapy: A Phase 2 Trial of Dose-Dense Methotrexate, Vinblastine, Doxorubicin, and Cisplatin with Bevacizumab in Urothelial Cancer. Eur. Urol. 2016, 69, 855–862. [Google Scholar]
- Seiler, R.; Ashab, H.A.D.; Erho, N.; van Rhijn, B.W.G.; Winters, B.; Douglas, J.; van Kessel, K.E.; Fransen van de Putte, E.E.; Sommerlad, M.; Wang, N.Q.; et al. Impact of Molecular Subtypes in Muscle-Invasive Bladder Cancer on Predicting Response and Survival after Neoadjuvant Chemotherapy. Eur. Urol. 2017, 72, 544–554. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGF-β Attenuates Tumour Response to PD-L1 Blockade by Contributing to Exclusion of T Cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Shariat, S.F.; Palapattu, G.S.; Karakiewicz, P.I.; Rogers, C.G.; Vazina, A.; Bastian, P.J.; Schoenberg, M.P.; Lerner, S.P.; Sagalowsky, A.I.; Lotan, Y. Discrepancy between Clinical and Pathologic Stage: Impact on Prognosis after Radical Cystectomy. Eur. Urol. 2007, 51, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Van Rhijn, B.W.G.; van der Kwast, T.H.; Kakiashvili, D.M.; Fleshner, N.E.; van der Aa, M.N.M.; Alkhateeb, S.; Bangma, C.H.; Jewett, M.A.S.; Zlotta, A.R. Pathological Stage Review Is Indicated in Primary PT1 Bladder Cancer. BJU Int. 2010, 106, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Svatek, R.S.; Shariat, S.F.; Novara, G.; Skinner, E.C.; Fradet, Y.; Bastian, P.J.; Kamat, A.M.; Kassouf, W.; Karakiewicz, P.I.; Fritsche, H.; et al. Discrepancy between Clinical and Pathological Stage: External Validation of the Impact on Prognosis in an International Radical Cystectomy Cohort: Stage discrepancy in urothelial carcinoma of the bladder. BJU Int. 2011, 107, 898–904. [Google Scholar] [CrossRef]
- Jones, P.A.; Droller, M.J. Pathways of Development and Progression in Bladder Cancer: New Correlations between Clinical Observations and Molecular Mechanisms. Semin. Urol. 1993, 11, 177–192. [Google Scholar]
- Cancer Genome Atlas Research Network. Comprehensive Molecular Characterization of Urothelial Bladder Carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Hedegaard, J.; Lamy, P.; Nordentoft, I.; Algaba, F.; Høyer, S.; Ulhøi, B.P.; Vang, S.; Reinert, T.; Hermann, G.G.; Mogensen, K.; et al. Comprehensive Transcriptional Analysis of Early-Stage Urothelial Carcinoma. Cancer Cell 2016, 30, 27–42. [Google Scholar] [CrossRef]
- Pietzak, E.J.; Bagrodia, A.; Cha, E.K.; Drill, E.N.; Iyer, G.; Isharwal, S.; Ostrovnaya, I.; Baez, P.; Li, Q.; Berger, M.F.; et al. Next-Generation Sequencing of Nonmuscle Invasive Bladder Cancer Reveals Potential Biomarkers and Rational Therapeutic Targets. Eur. Urol. 2017, 72, 952–959. [Google Scholar] [CrossRef]
- Sjödahl, G.; Lauss, M.; Lövgren, K.; Chebil, G.; Gudjonsson, S.; Veerla, S.; Patschan, O.; Aine, M.; Fernö, M.; Ringnér, M.; et al. A Molecular Taxonomy for Urothelial Carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 3377–3386. [Google Scholar] [CrossRef] [Green Version]
- van Rhijn, B.W.G.; Lurkin, I.; Radvanyi, F.; Kirkels, W.J.; van der Kwast, T.H.; Zwarthoff, E.C. The Fibroblast Growth Factor Receptor 3 (FGFR3) Mutation Is a Strong Indicator of Superficial Bladder Cancer with Low Recurrence Rate. Cancer Res. 2001, 61, 1265–1268. [Google Scholar] [PubMed]
- Zieger, K.; Marcussen, N.; Borre, M.; Ørntoft, T.F.; Dyrskjøt, L. Consistent Genomic Alterations in Carcinoma in Situ of the Urinary Bladder Confirm the Presence of Two Major Pathways in Bladder Cancer Development. Int. J. Cancer 2009, 125, 2095–2103. [Google Scholar] [CrossRef]
- Billerey, C.; Chopin, D.; Aubriot-Lorton, M.H.; Ricol, D.; Gil Diez de Medina, S.; Van Rhijn, B.; Bralet, M.P.; Lefrere-Belda, M.A.; Lahaye, J.B.; Abbou, C.C.; et al. Frequent FGFR3 Mutations in Papillary Non-Invasive Bladder (PTa) Tumors. Am. J. Pathol. 2001, 158, 1955–1959. [Google Scholar] [CrossRef] [Green Version]
- Hosen, I.; Rachakonda, P.S.; Heidenreich, B.; de Verdier, P.J.; Ryk, C.; Steineck, G.; Hemminki, K.; Kumar, R. Mutations in TERT Promoter and FGFR3 and Telomere Length in Bladder Cancer. Int. J. Cancer 2015, 137, 1621–1629. [Google Scholar] [CrossRef] [PubMed]
- Hussain, T.A.; Akhtar, M. Molecular Basis of Urinary Bladder Cancer. Adv. Anat. Pathol. 2013, 20, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Jebar, A.H.; Hurst, C.D.; Tomlinson, D.C.; Johnston, C.; Taylor, C.F.; Knowles, M.A. FGFR3 and Ras Gene Mutations Are Mutually Exclusive Genetic Events in Urothelial Cell Carcinoma. Oncogene 2005, 24, 5218–5225. [Google Scholar] [CrossRef] [Green Version]
- Hurst, C.D.; Knowles, M.A. Mutational Landscape of Non-Muscle-Invasive Bladder Cancer. Urol. Oncol. 2018. [Google Scholar] [CrossRef]
- Hurst, C.D.; Alder, O.; Platt, F.M.; Droop, A.; Stead, L.F.; Burns, J.E.; Burghel, G.J.; Jain, S.; Klimczak, L.J.; Lindsay, H.; et al. Genomic Subtypes of Non-Invasive Bladder Cancer with Distinct Metabolic Profile, Clinical Outcome and Female Gender Bias in KDM6A Mutation Frequency. Cancer Cell 2017, 32, 701–715. [Google Scholar] [CrossRef]
- López-Knowles, E.; Hernández, S.; Malats, N.; Kogevinas, M.; Lloreta, J.; Carrato, A.; Tardón, A.; Serra, C.; Real, F.X. PIK3CA Mutations Are an Early Genetic Alteration Associated with FGFR3 Mutations in Superficial Papillary Bladder Tumors. Cancer Res. 2006, 66, 7401–7404. [Google Scholar] [CrossRef] [Green Version]
- Platt, F.M.; Hurst, C.D.; Taylor, C.F.; Gregory, W.M.; Harnden, P.; Knowles, M.A. Spectrum of Phosphatidylinositol 3-Kinase Pathway Gene Alterations in Bladder Cancer. Clin. Cancer Res. 2009, 15, 6008–6017. [Google Scholar] [CrossRef] [Green Version]
- Meeks, J.J.; Carneiro, B.A.; Pai, S.G.; Oberlin, D.T.; Rademaker, A.; Fedorchak, K.; Balasubramanian, S.; Elvin, J.; Beaubier, N.; Giles, F.J. Genomic Characterization of High-Risk Non-Muscle Invasive Bladder Cancer. Oncotarget 2016, 7, 75176–75184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, V.G.; Munera-Maravilla, E.; Bernardini, A.; Rubio, C.; Suarez-Cabrera, C.; Segovia, C.; Lodewijk, I.; Dueñas, M.; Martínez-Fernández, M.; Paramio, J.M. Epigenetics of Bladder Cancer: Where Biomarkers and Therapeutic Targets Meet. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramakrishnan, S.; Granger, V.; Rak, M.; Hu, Q.; Attwood, K.; Aquila, L.; Krishnan, N.; Osiecki, R.; Azabdaftari, G.; Guru, K.; et al. Inhibition of EZH2 Induces NK Cell-Mediated Differentiation and Death in Muscle-Invasive Bladder Cancer. Cell Death Differ. 2019, 26, 2100–2114. [Google Scholar] [CrossRef] [PubMed]
- Solomon, D.A.; Kim, J.-S.; Bondaruk, J.; Shariat, S.F.; Wang, Z.-F.; Elkahloun, A.G.; Ozawa, T.; Gerard, J.; Zhuang, D.; Zhang, S.; et al. Frequent Truncating Mutations of STAG2 in Bladder Cancer. Nat. Genet. 2013, 45, 1428–1430. [Google Scholar] [CrossRef]
- Aboulkassim, T.O.; LaRue, H.; Lemieux, P.; Rousseau, F.; Fradet, Y. Alteration of the PATCHED Locus in Superficial Bladder Cancer. Oncogene 2003, 22, 2967–2971. [Google Scholar] [CrossRef] [Green Version]
- Knowles, M.A.; Habuchi, T.; Kennedy, W.; Cuthbert-Heavens, D. Mutation Spectrum of the 9q34 Tuberous Sclerosis Gene TSC1 in Transitional Cell Carcinoma of the Bladder. Cancer Res. 2003, 63, 7652–7656. [Google Scholar]
- Blaveri, E.; Simko, J.P.; Korkola, J.E.; Brewer, J.L.; Baehner, F.; Mehta, K.; DeVries, S.; Koppie, T.; Pejavar, S.; Carroll, P.; et al. Bladder Cancer Outcome and Subtype Classification by Gene Expression. Clin. Cancer Res. 2005, 11, 4044–4055. [Google Scholar] [CrossRef] [Green Version]
- Williamson, M.P.; Elder, P.A.; Shaw, M.E.; Devlin, J.; Knowles, M.A. P16 (CDKN2) Is a Major Deletion Target at 9p21 in Bladder Cancer. Hum. Mol. Genet. 1995, 4, 1569–1577. [Google Scholar] [CrossRef]
- Bartoletti, R.; Cai, T.; Nesi, G.; Roberta Girardi, L.; Baroni, G.; Dal Canto, M. Loss of P16 Expression and Chromosome 9p21 LOH in Predicting Outcome of Patients Affected by Superficial Bladder Cancer. J. Surg. Res. 2007, 143, 422–427. [Google Scholar] [CrossRef]
- Rebouissou, S.; Bernard-Pierrot, I.; de Reyniès, A.; Lepage, M.-L.; Krucker, C.; Chapeaublanc, E.; Hérault, A.; Kamoun, A.; Caillault, A.; Letouzé, E.; et al. EGFR as a Potential Therapeutic Target for a Subset of Muscle-Invasive Bladder Cancers Presenting a Basal-like Phenotype. Sci. Transl. Med. 2014, 6, 244ra91. [Google Scholar] [CrossRef]
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017, 171, 540–556.e25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baud, E.; Catilina, P.; Bignon, Y.J. P16 Involvement in Primary Bladder Tumors: Analysis of Deletions and Mutations. Int. J. Oncol. 1999, 14, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Morrison, C.D.; Liu, P.; Woloszynska-Read, A.; Zhang, J.; Luo, W.; Qin, M.; Bshara, W.; Conroy, J.M.; Sabatini, L.; Vedell, P.; et al. Whole-Genome Sequencing Identifies Genomic Heterogeneity at a Nucleotide and Chromosomal Level in Bladder Cancer. Proc. Natl. Acad. Sci. USA 2014, 111, E672–E681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shariat, S.F.; Tokunaga, H.; Zhou, J.; Kim, J.; Ayala, G.E.; Benedict, W.F.; Lerner, S.P. P53, P21, PRB, and P16 Expression Predict Clinical Outcome in Cystectomy With Bladder Cancer. J. Clin. Oncol. 2004, 22, 1014–1024. [Google Scholar] [CrossRef] [PubMed]
- Kamoun, A.; de Reyniès, A.; Allory, Y.; Sjödahl, G.; Robertson, A.G.; Seiler, R.; Hoadley, K.A.; Groeneveld, C.S.; Al-Ahmadie, H.; Choi, W.; et al. A Consensus Molecular Classification of Muscle-Invasive Bladder Cancer. Eur. Urol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Ross, J.S.; Wang, K.; Al-Rohil, R.N.; Nazeer, T.; Sheehan, C.E.; Otto, G.A.; He, J.; Palmer, G.; Yelensky, R.; Lipson, D.; et al. Advanced Urothelial Carcinoma: Next-Generation Sequencing Reveals Diverse Genomic Alterations and Targets of Therapy. Mod. Pathol. Off. J. U. S. Can. Acad. Pathol. Inc. 2014, 27, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Sjödahl, G.; Eriksson, P.; Liedberg, F.; Höglund, M. Molecular Classification of Urothelial Carcinoma: Global MRNA Classification versus Tumour-Cell Phenotype Classification. J. Pathol. 2017, 242, 113–125. [Google Scholar] [CrossRef]
- Cordon-Cardo, C.; Zhang, Z.-F.; Dalbagni, G.; Drobnjak, M.; Charytonowicz, E.; Hu, S.-X.; Xu, H.-J.; Reuter, V.E.; Benedict, W.F. Cooperative Effects of P53 and PRB Alterations in Primary Superficial Bladder Tumors. Cancer Res. 1997, 57, 1217–1221. [Google Scholar]
- Grossman, H.B.; Liebert, M.; Antelo, M.; Dinney, C.P.; Hu, S.X.; Palmer, J.L.; Benedict, W.F. P53 and RB Expression Predict Progression in T1 Bladder Cancer. Clin. Cancer Res. 1998, 4, 829–834. [Google Scholar]
- Sherr, C.J.; McCormick, F. The RB and P53 Pathways in Cancer. Cancer Cell 2002, 2, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Esteller, M. Epigenetics in Cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Mouw, K.W.; Polak, P.; Braunstein, L.Z.; Kamburov, A.; Kwiatkowski, D.J.; Rosenberg, J.E.; Van Allen, E.M.; D’Andrea, A.; Getz, G. Somatic ERCC2 Mutations Are Associated with a Distinct Genomic Signature in Urothelial Tumors. Nat. Genet. 2016, 48, 600–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Allen, E.M.; Mouw, K.W.; Kim, P.; Iyer, G.; Wagle, N.; Al-Ahmadie, H.; Zhu, C.; Ostrovnaya, I.; Kryukov, G.V.; O’Connor, K.W.; et al. Somatic ERCC2 Mutations Correlate with Cisplatin Sensitivity in Muscle-Invasive Urothelial Carcinoma. Cancer Discov. 2014, 4, 1140–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, G.; Sun, X.; Chen, C.; Wu, S.; Huang, P.; Li, Z.; Dean, M.; Huang, Y.; Jia, W.; Zhou, Q.; et al. Whole-Genome and Whole-Exome Sequencing of Bladder Cancer Identifies Frequent Alterations in Genes Involved in Sister Chromatid Cohesion and Segregation. Nat. Genet. 2013, 45, 1459–1463. [Google Scholar] [CrossRef]
- Carlsson, J.; Wester, K.; De La Torre, M.; Malmström, P.-U.; Gårdmark, T. EGFR-Expression in Primary Urinary Bladder Cancer and Corresponding Metastases and the Relation to HER2-Expression. On the Possibility to Target These Receptors with Radionuclides. Radiol. Oncol. 2015, 49, 50–58. [Google Scholar] [CrossRef] [Green Version]
- Gildea, J.J.; Harding, M.A.; Seraj, M.J.; Gulding, K.M.; Theodorescu, D. The Role of Ral A in Epidermal Growth Factor Receptor-Regulated Cell Motility. Cancer Res. 2002, 62, 982–985. [Google Scholar]
- Seshacharyulu, P.; Ponnusamy, M.P.; Haridas, D.; Jain, M.; Ganti, A.p.a.r.K.; Batra, S.K. Targeting the EGFR Signaling Pathway in Cancer Therapy. Expert Opin. Ther. Targets 2012, 16, 15–31. [Google Scholar] [CrossRef] [Green Version]
- Theodorescu, D.; Laderoute, K.R.; Gulding, K.M. Epidermal Growth Factor Receptor-Regulated Human Bladder Cancer Motility Is in Part a Phosphatidylinositol 3-Kinase-Mediated Process. Cell Growth Differ. 1998, 9, 919. [Google Scholar]
- Memon, A.A.; Sorensen, B.S.; Melgard, P.; Fokdal, L.; Thykjaer, T.; Nexo, E. Expression of HER3, HER4 and Their Ligand Heregulin-4 Is Associated with Better Survival in Bladder Cancer Patients. Br. J. Cancer 2004, 91, 2034–2041. [Google Scholar] [CrossRef] [Green Version]
- Coogan, C.L.; Estrada, C.R.; Kapur, S.; Bloom, K.J. HER-2/Neu Protein Overexpression and Gene Amplification in Human Transitional Cell Carcinoma of the Bladder. Urology 2004, 63, 786–790. [Google Scholar] [CrossRef]
- Maida, F.D.; Mari, A.; Gesolfo, C.S.; Cangemi, A.; Allegro, R.; Sforza, S.; Cocci, A.; Tellini, R.; Masieri, L.; Russo, A.; et al. Epidermal Growth Factor Receptor (EGFR) Cell Expression During Adjuvant Treatment After Transurethral Resection for Non-Muscle-Invasive Bladder Cancer: A New Potential Tool to Identify Patients at Higher Risk of Disease Progression. Clin. Genitourin. Cancer 2019, 17, e751–e758. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, A.; Rotzer, D.; Seiler, R.; Studer, U.E.; Thalmann, G.N. Her2 Amplification Is Significantly More Frequent in Lymph Node Metastases from Urothelial Bladder Cancer than in the Primary Tumours. Eur. Urol. 2011, 60, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.A.; Danielpour, D. MTOR Inhibition as a Therapeutic Strategy in the Management of Urologic Malignancies. Mol. Cancer Ther. 2008, 7, 1347–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gildea, J.J.; Herlevsen, M.; Harding, M.A.; Gulding, K.M.; Moskaluk, C.A.; Frierson, H.F.; Theodorescu, D. PTEN Can Inhibit in Vitro Organotypic and in Vivo Orthotopic Invasion of Human Bladder Cancer Cells Even in the Absence of Its Lipid Phosphatase Activity. Oncogene 2004, 23, 6788–6797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aveyard, J.S.; Skilleter, A.; Habuchi, T.; Knowles, M.A. Somatic Mutation of PTEN in Bladder Carcinoma. Br. J. Cancer 1999, 80, 904–908. [Google Scholar] [CrossRef] [Green Version]
- Puzio-Kuter, A.M.; Castillo-Martin, M.; Kinkade, C.W.; Wang, X.; Shen, T.H.; Matos, T.; Shen, M.M.; Cordon-Cardo, C.; Abate-Shen, C. Inactivation of P53 and Pten Promotes Invasive Bladder Cancer. Genes Dev. 2009, 23, 675–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, Q.; Nikolos, F.; Chen, F.; Tramel, Z.; Lee, Y.-C.; Hayashi, K.; Xiao, J.; Shen, J.; Chan, K.S. Prognostic Power of a Tumor Differentiation Gene Signature for Bladder Urothelial Carcinomas. JNCI J. Natl. Cancer Inst. 2018, 110, 448–459. [Google Scholar] [CrossRef]
- Choi, W.; Czerniak, B.; Ochoa, A.; Su, X.; Siefker-Radtke, A.; Dinney, C.; McConkey, D.J. Intrinsic Basal and Luminal Subtypes of Muscle-Invasive Bladder Cancer. Nat. Rev. Urol. 2014, 11, 400–410. [Google Scholar] [CrossRef]
- Choi, W.; Porten, S.; Kim, S.; Willis, D.; Plimack, E.R.; Hoffman-Censits, J.; Roth, B.; Cheng, T.; Tran, M.; Lee, I.-L.; et al. Identification of Distinct Basal and Luminal Subtypes of Muscle-Invasive Bladder Cancer with Different Sensitivities to Frontline Chemotherapy. Cancer Cell 2014, 25, 152–165. [Google Scholar] [CrossRef] [Green Version]
- Damrauer, J.S.; Hoadley, K.A.; Chism, D.D.; Fan, C.; Tiganelli, C.J.; Wobker, S.E.; Yeh, J.J.; Milowsky, M.I.; Iyer, G.; Parker, J.S.; et al. Intrinsic Subtypes of High-Grade Bladder Cancer Reflect the Hallmarks of Breast Cancer Biology. Proc. Natl. Acad. Sci. USA 2014, 111, 3110–3115. [Google Scholar] [CrossRef] [Green Version]
- Kardos, J.; Chai, S.; Mose, L.E.; Selitsky, S.R.; Krishnan, B.; Saito, R.; Iglesia, M.D.; Milowsky, M.I.; Parker, J.S.; Kim, W.Y.; et al. Claudin-Low Bladder Tumors Are Immune Infiltrated and Actively Immune Suppressed. JCI Insight 1 2016, 1, e85902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzouka, N.; Eriksson, P.; Rovira, C.; Liedberg, F.; Sjödahl, G.; Höglund, M. A Validation and Extended Description of the Lund Taxonomy for Urothelial Carcinoma Using the TCGA Cohort. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nag, S.; Qin, J.; Srivenugopal, K.S.; Wang, M.; Zhang, R. The MDM2-P53 Pathway Revisited. J. Biomed. Res. 2013, 27, 254–271. [Google Scholar] [PubMed]
- He, F.; Mo, L.; Zheng, X.-Y.; Hu, C.; Lepor, H.; Lee, E.Y.-H.P.; Sun, T.-T.; Wu, X.-R. Deficiency of PRb Family Proteins and P53 in Invasive Urothelial Tumorigenesis. Cancer Res. 2009, 69, 9413–9421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McConkey, D.J.; Choi, W.; Marquis, L.; Martin, F.; Williams, M.B.; Shah, J.; Svatek, R.; Das, A.; Adam, L.; Kamat, A.; et al. Role of Epithelial-to-Mesenchymal Transition (EMT) in Drug Sensitivity and Metastasis in Bladder Cancer. Cancer Metastasis Rev. 2009, 28, 335–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, H.-W.; Hsu, S.-C.; Xia, W.; Cao, X.; Shih, J.-Y.; Wei, Y.; Abbruzzese, J.L.; Hortobagyi, G.N.; Hung, M.-C. Epidermal Growth Factor Receptor Cooperates with Signal Transducer and Activator of Transcription 3 to Induce Epithelial-Mesenchymal Transition in Cancer Cells via Up-Regulation of TWIST Gene Expression. Cancer Res. 2007, 67, 9066–9076. [Google Scholar] [CrossRef] [Green Version]
- Kurzrock, E.A.; Lieu, D.K.; deGraffenried, L.A.; Chan, C.W.; Isseroff, R.R. Label-Retaining Cells of the Bladder: Candidate Urothelial Stem Cells. Am. J. Physiol.-Ren. Physiol. 2008, 294, F1415–F1421. [Google Scholar] [CrossRef]
- Choi, W.; Shah, J.B.; Tran, M.; Svatek, R.; Marquis, L.; Lee, I.-L.; Yu, D.; Adam, L.; Wen, S.; Shen, Y.; et al. P63 Expression Defines a Lethal Subset of Muscle-Invasive Bladder Cancers. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [Green Version]
- Karni-Schmidt, O.; Castillo-Martin, M.; HuaiShen, T.; Gladoun, N.; Domingo-Domenech, J.; Sanchez-Carbayo, M.; Li, Y.; Lowe, S.; Prives, C.; Cordon-Cardo, C. Distinct Expression Profiles of P63 Variants during Urothelial Development and Bladder Cancer Progression. Am. J. Pathol. 2011, 178, 1350–1360. [Google Scholar] [CrossRef]
- Chu, W.-K.; Dai, P.-M.; Li, H.-L.; Chen, J.-K. Transcriptional Activity of the ΔNp63 Promoter Is Regulated by STAT3. J. Biol. Chem. 2008, 283, 7328–7337. [Google Scholar] [CrossRef] [Green Version]
- Kollberg, P.; Chebil, G.; Eriksson, P.; Sjödahl, G.; Liedberg, F. Molecular Subtypes Applied to a Population-Based Modern Cystectomy Series Do Not Predict Cancer-Specific Survival. Urol. Oncol. Semin. Orig. Investig. 2019, 37, 791–799. [Google Scholar] [CrossRef] [PubMed]
- van Rhijn BW, G.; van der Kwast, T.H.; Vis, A.N.; Kirkels, W.J.; Boevé, E.R.; Jöbsis, A.C.; Zwarthoff, E.C. FGFR3 and P53 Characterize Alternative Genetic Pathways in the Pathogenesis of Urothelial Cell Carcinoma. Cancer Res. 2004, 64, 1911–1914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomlinson, D.C.; Baldo, O.; Harnden, P.; Knowles, M.A. FGFR3 Protein Expression and Its Relationship to Mutation Status and Prognostic Variables in Bladder Cancer. J. Pathol. 2007, 213, 91–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Rhijn Bas, W.G.; van der Kwast Theo, H.; Liu, L.; Fleshner Neil, E.; Bostrom Peter, J.; Vis André, N.; Alkhateeb Sultan, S.; Bangma Chris, H.; Jewett Michael, A.S.; Zwarthoff Ellen, C.; et al. The FGFR3 Mutation Is Related to Favorable PT1 Bladder Cancer. J. Urol. 2012, 187, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.G.; Pant, V.; Li, Q.; Chang, L.L.; Quintás-Cardama, A.; Garza, D.; Tavana, O.; Yang, P.; Manshouri, T.; Li, Y.; et al. P53 Mediated Senescence Impairs the Apoptotic Response to Chemotherapy and Clinical Outcome in Breast Cancer. Cancer Cell 2012, 21, 793–806. [Google Scholar] [CrossRef] [Green Version]
- López-Knowles, E.; Hernández, S.; Kogevinas, M.; Lloreta, J.; Amorós, A.; Tardón, A.; Carrato, A.; Kishore, S.; Serra, C.; Malats, N.; et al. The P53 Pathway and Outcome among Patients with T1G3 Bladder Tumors. Clin. Cancer Res. 2006, 12, 6029–6036. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Smith, B.A.; Balanis, N.G.; Tsai, B.L.; Nguyen, K.; Cheng, M.W.; Obusan, M.B.; Esedebe, F.N.; Patel, S.J.; Zhang, H.; et al. A Genetically Defined Disease Model Reveals That Urothelial Cells Can Initiate Divergent Bladder Cancer Phenotypes. Proc. Natl. Acad. Sci. USA 2020, 117, 563–572. [Google Scholar] [CrossRef] [Green Version]
- Ghervan, L.; Zaharie, A.; Ene, B.; Elec, F.I. Small-Cell Carcinoma of the Urinary Bladder: Where Do We Stand? Clujul Med. 2017, 90, 13–17. [Google Scholar] [CrossRef]
- Chang, M.T.; Penson, A.; Desai, N.B.; Socci, N.D.; Shen, R.; Seshan, V.E.; Kundra, R.; Abeshouse, A.; Viale, A.; Cha, E.K.; et al. Small Cell Carcinomas of the Bladder and Lung Are Characterized by a Convergent but Distinct Pathogenesis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 1965–1973. [Google Scholar] [CrossRef] [Green Version]
- Nadal, R.; Schweizer, M.; Kryvenko, O.N.; Epstein, J.I.; Eisenberger, M.A. Small Cell Carcinoma of the Prostate. Nat. Rev. Urol. 2014, 11, 213–219. [Google Scholar] [CrossRef]
- Seiler, R.; Gibb, E.A.; Wang, N.Q.; Oo, H.Z.; Lam, H.-M.; van Kessel, K.E.; Voskuilen, C.S.; Winters, B.; Erho, N.; Takhar, M.M.; et al. Divergent Biological Response to Neoadjuvant Chemotherapy in Muscle-Invasive Bladder Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 5082–5093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Taylor, J.; Eustace, A.; Irlam, J.J.; Denley, H.; Hoskin, P.J.; Alsner, J.; Buffa, F.M.; Harris, A.L.; Choudhury, A. Gene Signature for Selecting Benefit from Hypoxia Modification of Radiotherapy for High-Risk Bladder Cancer Patients. Clin. Cancer Res. 2017, 23, 4761–4768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efstathiou, J.A.; Mouw, K.W.; Gibb, E.A.; Liu, Y.; Wu, C.-L.; Drumm, M.R.; da Costa, J.B.; du Plessis, M.; Wang, N.Q.; Davicioni, E.; et al. Impact of Immune and Stromal Infiltration on Outcomes Following Bladder-Sparing Trimodality Therapy for Muscle-Invasive Bladder Cancer. Eur. Urol. 2019, 76, 59–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Necchi, A.; Raggi, D.; Gallina, A.; Ross, J.S.; Farè, E.; Giannatempo, P.; Marandino, L.; Colecchia, M.; Lucianò, R.; Bianchi, M.; et al. Impact of Molecular Subtyping and Immune Infiltration on Pathological Response and Outcome Following Neoadjuvant Pembrolizumab in Muscle-Invasive Bladder Cancer. Eur. Urol. 2020, 77, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kwiatkowski, D.; McConkey, D.J.; Meeks, J.J.; Freeman, S.S.; Bellmunt, J.; Getz, G.; Lerner, S.P. The Cancer Genome Atlas Expression Subtypes Stratify Response to Checkpoint Inhibition in Advanced Urothelial Cancer and Identify a Subset of Patients with High Survival Probability. Eur. Urol. 2019, 75, 961–964. [Google Scholar] [CrossRef]
- Dutta, P.R.; Maity, A. Cellular Responses to EGFR Inhibitors and Their Relevance to Cancer Therapy. Cancer Lett. 2007, 254, 165–177. [Google Scholar] [CrossRef] [Green Version]
- Powles, T.; Huddart, R.A.; Elliott, T.; Sarker, S.-J.; Ackerman, C.; Jones, R.; Hussain, S.; Crabb, S.; Jagdev, S.; Chester, J.; et al. Phase III, Double-Blind, Randomized Trial That Compared Maintenance Lapatinib Versus Placebo After First-Line Chemotherapy in Patients With Human Epidermal Growth Factor Receptor 1/2–Positive Metastatic Bladder Cancer. J. Clin. Oncol. 2016, 35, 48–55. [Google Scholar] [CrossRef]
- Hussain, M.; Daignault, S.; Agarwal, N.; Grivas, P.D.; Siefker-Radtke, A.O.; Puzanov, I.; MacVicar, G.R.; Levine, E.G.; Srinivas, S.; Twardowski, P.; et al. Randomized Phase 2 Trial of Gemcitabine/Cisplatin with or without Cetuximab in Patients with Advanced Urothelial Carcinoma. Cancer 2014, 120, 2684–2693. [Google Scholar] [CrossRef] [Green Version]
- Stephenson, J.J.; Gregory, C.; Burris, H.; Larson, T.; Verma, U.; Cohn, A.; Crawford, J.; Cohen, R.B.; Martin, J.; Lum, P.; et al. An Open-Label Clinical Trial Evaluating Safety and Pharmacokinetics of Two Dosing Schedules of Panitumumab in Patients with Solid Tumors. Clin. Colorectal Cancer 2009, 8, 29–37. [Google Scholar] [CrossRef]
- Wong, Y.-N.; Litwin, S.; Vaughn, D.; Cohen, S.; Plimack, E.R.; Lee, J.; Song, W.; Dabrow, M.; Brody, M.; Tuttle, H.; et al. Phase II Trial of Cetuximab With or Without Paclitaxel in Patients With Advanced Urothelial Tract Carcinoma. J. Clin. Oncol. 2012, 30, 3545–3551. [Google Scholar] [CrossRef] [Green Version]
- Pruthi, R.S.; Nielsen, M.; Heathcote, S.; Wallen, E.M.; Rathmell, W.K.; Godley, P.; Whang, Y.; Fielding, J.; Schultz, H.; Grigson, G.; et al. A Phase II Trial of Neoadjuvant Erlotinib in Patients with Muscle-Invasive Bladder Cancer Undergoing Radical Cystectomy: Clinical and Pathological Results. BJU Int. 2010, 106, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Petrylak, D.P.; Tangen, C.M.; Van Veldhuizen Jr, P.J.; Goodwin, J.W.; Twardowski, P.W.; Atkins, J.N.; Kakhil, S.R.; Lange, M.K.; Mansukhani, M.; Crawford, E.D. Results of the Southwest Oncology Group Phase II Evaluation (Study S0031) of ZD1839 for Advanced Transitional Cell Carcinoma of the Urothelium. BJU Int. 2010, 105, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Boehringer Ingelheim. LUX-Bladder 1: Phase II Open Label Single Arm Exploratory Trial of Oral Afatinib Monotherapy Following Platinum Failure for Patients with Advanced/Metastatic Urothelial Tract Carcinoma with Genetic Alterations in ERBB Receptors.; Boehringer Ingelheim: Ingelheim am Rhein, Germany, 2019; Clinical trial registration NCT02780687; clinicaltrials.gov. [Google Scholar]
- Huang, L.; Fu, L. Mechanisms of Resistance to EGFR Tyrosine Kinase Inhibitors. Acta Pharm. Sin. B 2015, 5, 390–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douillard, J.-Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab–FOLFOX4 Treatment and RAS Mutations in Colorectal Cancer. Available online: https://www.nejm.org/doi/10.1056/NEJMoa1305275?url_ver=Z39.88-2003&rfr_id=ori%3Arid%3Acrossref.org&rfr_dat=cr_pub%3Dwww.ncbi.nlm.nih.gov (accessed on 16 April 2020).
- Oudard, S.; Culine, S.; Vano, Y.; Goldwasser, F.; Théodore, C.; Nguyen, T.; Voog, E.; Banu, E.; Vieillefond, A.; Priou, F.; et al. Multicentre Randomised Phase II Trial of Gemcitabine+platinum, with or without Trastuzumab, in Advanced or Metastatic Urothelial Carcinoma Overexpressing Her2. Eur. J. Cancer 2015, 51, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Kiss, B.; Wyatt, A.W.; Douglas, J.; Skuginna, V.; Mo, F.; Anderson, S.; Rotzer, D.; Fleischmann, A.; Genitsch, V.; Hayashi, T.; et al. Her2 Alterations in Muscle-Invasive Bladder Cancer: Patient Selection beyond Protein Expression for Targeted Therapy. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Koshkin, V.S.; O’Donnell, P.; Yu, E.Y.; Grivas, P. Systematic Review: Targeting HER2 in Bladder Cancer. Bladder Cancer 2019, 5, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Thomsen MB, H.; Nordentoft, I.; Lamy, P.; Vang, S.; Reinert, L.; Mapendano, C.K.; Høyer, S.; Ørntoft, T.F.; Jensen, J.B.; Dyrskjøt, L. Comprehensive Multiregional Analysis of Molecular Heterogeneity in Bladder Cancer. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Genitsch, V.; Kollár, A.; Vandekerkhove, G.; Blarer, J.; Furrer, M.; Annala, M.; Herberts, C.; Pycha, A.; de Jong, J.J.; Liu, Y.; et al. Morphologic and Genomic Characterization of Urothelial to Sarcomatoid Transition in Muscle-Invasive Bladder Cancer†. Urol. Oncol. Semin. Orig. Investig. 2019, 37, 826–836. [Google Scholar] [CrossRef]
- Sjödahl, G.; Jackson, C.L.; Bartlett, J.M.; Siemens, D.R.; Berman, D.M. Molecular Profiling in Muscle-Invasive Bladder Cancer: More than the Sum of Its Parts. J. Pathol. 2019, 247, 563–573. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| NMIBC Subtypes | Differentiation | Oncogenic Mechanism | Molecular Features | Stage/Grade | Associated Risk |
|---|---|---|---|---|---|
| Class I | Luminal | FGFR3 mutations | ERBB3 overexpression High RAS/RAF/MAPK pathway activity Early cell cycle regulators (CCDN1 amplification) | Low stage and grade | High risk of reoccurrence |
| Class II | Luminal | HER2 amplification | TP53/RB1 loss ERCC2 mutations Late cell cycle regulators EMT transcript factors APOBEC mutation signatures | High stage and grade | High risk of progression |
| Class III | Basal | FGFR3 mutations | Dormant tumor state of NMIBC that switch to Class II upon progression | High stage and grade | High risk of progression |
| MIBC Subtypes | Differentiation | Oncogenic Mechanism | Molecular Features | Stage/Grade | Subsets |
|---|---|---|---|---|---|
| LumP | Luminal | FGFR3 mutations CDKN2A deletions | High RAS/RAF/MAPK pathway activity KDM6A mutations STAG2 mutations | T2+ | p53-like: WT p53 gene expression signature |
| LumNS | Luminal | PPARG mutations | ELF3 mutations Elevated stromal and moderate immune infiltration | T2+ | Luminal-infiltrated |
| LumU | Luminal | HER2 amplification Genomic instability | TP53 loss ERCC2 mutations High cell cycle activity APOBEC+ PPARG mutations | T2+ | |
| Ba/Sq | Basal and squamous | EGFR mutations | TP53 and/or RB1 loss STAT3 regulon activation Elevated HIF1A activity | T3/T4 | Claudin-low: Decreased expression of claudin-3 and claudin-4 Enrichment for TIC Overexpression of EMT transcript factors Stroma and immune infiltration High risk of metastatic disease |
| Stroma-rich | Basal and luminal | Elevated stroma and immune cells infiltration | |||
| NE-like | Neuroendocrine | TP53 and/or RB1 loss | High cell cycle activity Urothelial transdifferentiation |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minoli, M.; Kiener, M.; Thalmann, G.N.; Kruithof-de Julio, M.; Seiler, R. Evolution of Urothelial Bladder Cancer in the Context of Molecular Classifications. Int. J. Mol. Sci. 2020, 21, 5670. https://doi.org/10.3390/ijms21165670
Minoli M, Kiener M, Thalmann GN, Kruithof-de Julio M, Seiler R. Evolution of Urothelial Bladder Cancer in the Context of Molecular Classifications. International Journal of Molecular Sciences. 2020; 21(16):5670. https://doi.org/10.3390/ijms21165670
Chicago/Turabian StyleMinoli, Martina, Mirjam Kiener, George N. Thalmann, Marianna Kruithof-de Julio, and Roland Seiler. 2020. "Evolution of Urothelial Bladder Cancer in the Context of Molecular Classifications" International Journal of Molecular Sciences 21, no. 16: 5670. https://doi.org/10.3390/ijms21165670