
Defect Formation, T-Atom Substitution and Adsorption of Guest Molecules in MSE-Type Zeolite Framework—DFT Modeling

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results and Discussion
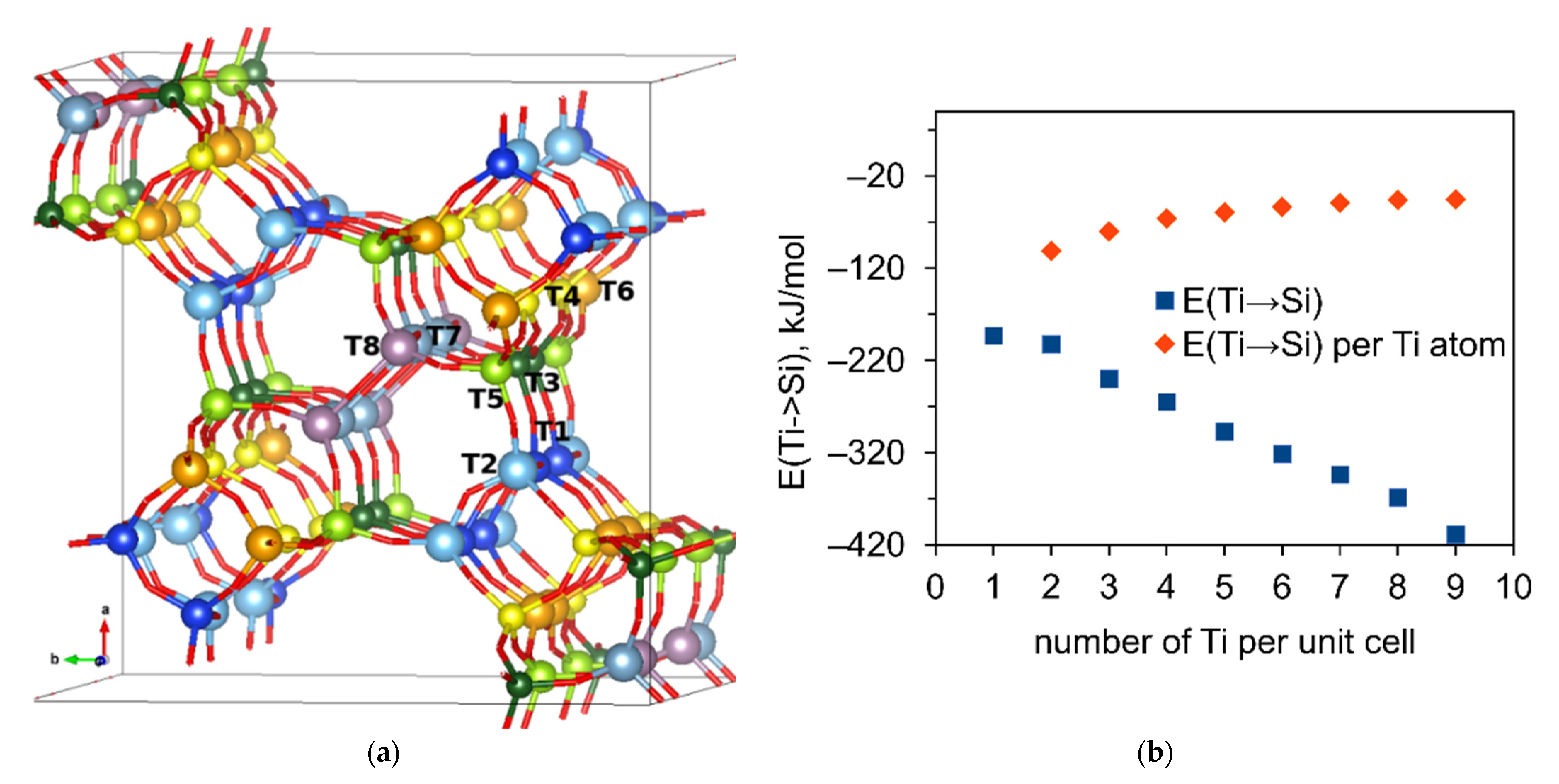
2.1. Formation of Si Defects in MSE-Type Framework
2.2. Substitution of Si by Ti or Al
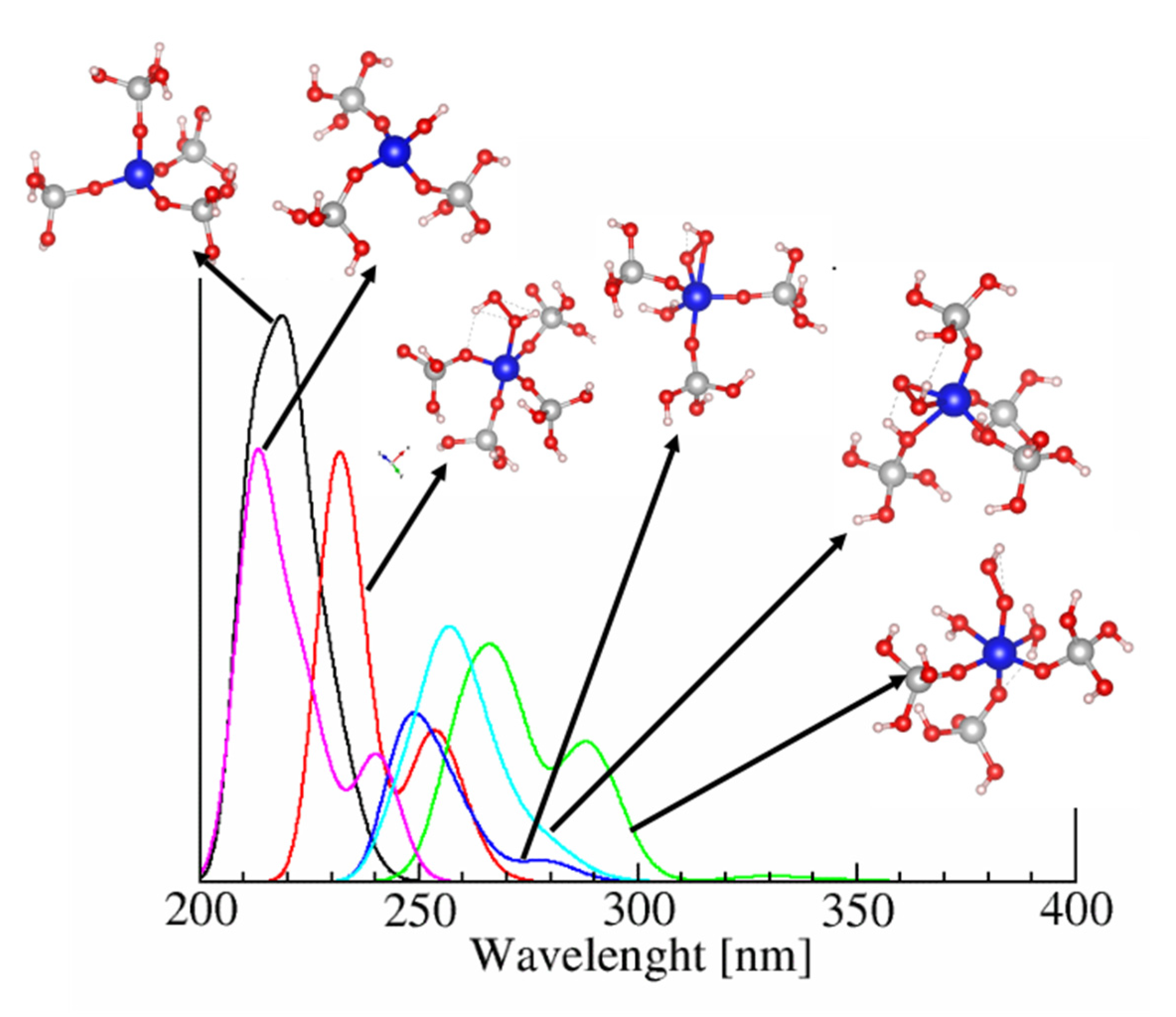
2.3. Adsorption Complexes of Pyridine and Acetonitrile on Ti Centers
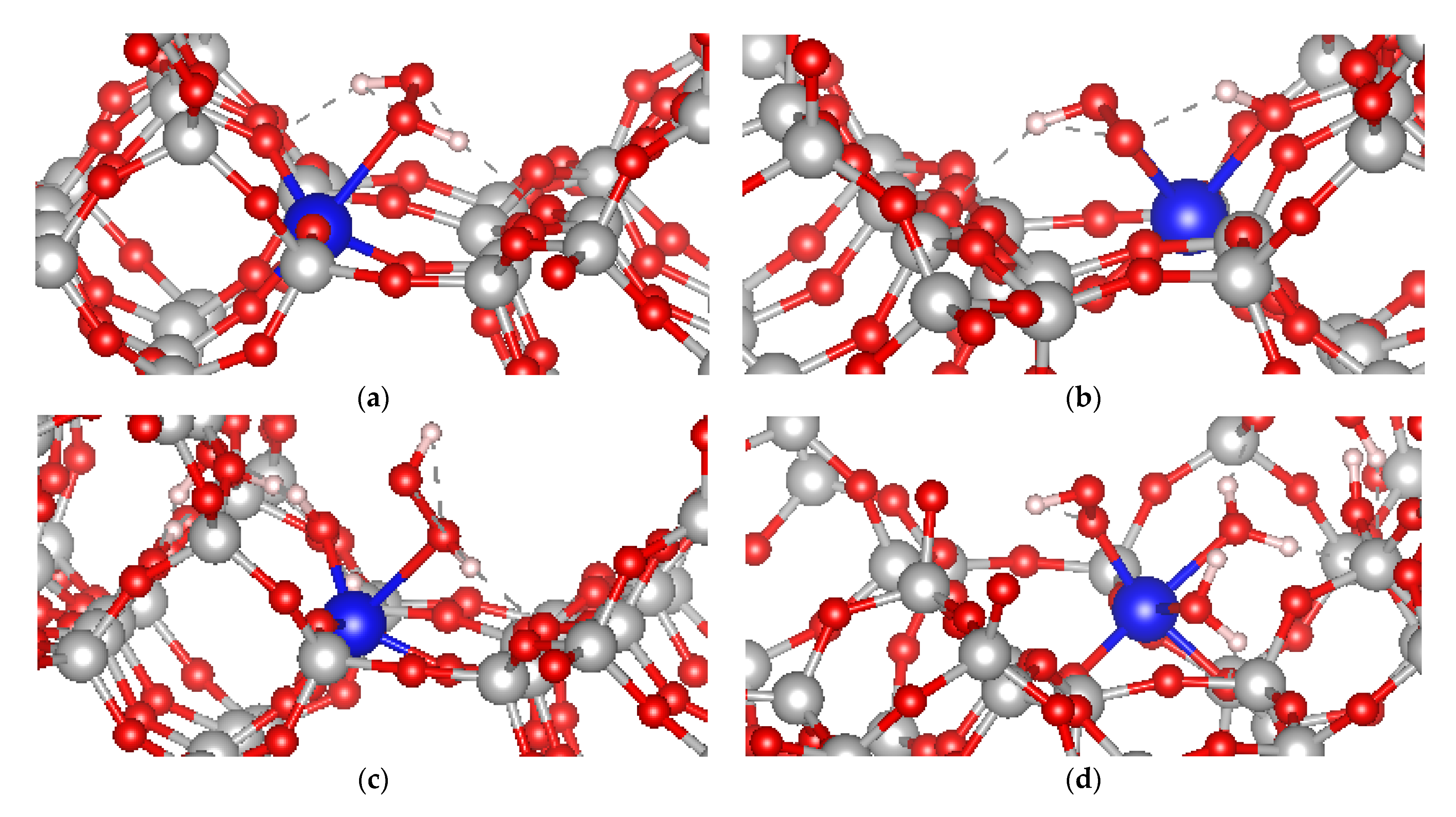
2.4. Adsorption of Hydrogen Peroxide at Ti in Ti-MSE in Presence or Absence of Si Vacancy
3. Discussion
4. Computational Details
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Clark, J.H. Green chemistry: Challenges and opportunities. Green Chem. 1999, 1, 1–8. [Google Scholar] [CrossRef]
- Notari, B. Microporous Crystalline Titanium Silicates. Adv. Catal. 1996, 41, 253–334. [Google Scholar] [CrossRef]
- Tatsumi, T. Metallozeolites and applications in catalysis. Curr. Opin. Solid State Mater. Sci. 1997, 2, 76–83. [Google Scholar] [CrossRef]
- Arends, I.W.C.E.; Sheldon, R.A. Activities and stabilities of heterogeneous catalysts in selective liquid phase oxidations: Recent developments. Appl. Catal. A Gen. 2001, 212, 175–187. [Google Scholar] [CrossRef]
- Ratnasamy, P.; Srinivas, D.; Knözinger, H. Active Sites and Reactive Intermediates in Titanium Silicate Molecular Sieves. Adv. Catal. 2004, 35, 48. [Google Scholar]
- Wu, P.; Tatsumi, T. A new generation of titanosilicate catalyst: Preparation and application to liquid-phase epoxidation of alkenes. Catal. Surv. Asia 2004, 8, 137–148. [Google Scholar] [CrossRef]
- Taramasso, M.; Milanese, S.; Perego, G.; Milan; Notari, B. Preparation of Porous Crystalline Synthetic Material Comprised of Silicon and Titanium Oxides. U.S. Patent No. 4,410,501, 18 October 1983. [Google Scholar]
- Vayssilov, G.N. Structural and physicochemical features of titanium silicalites. Catal. Rev. Sci. Eng. 1997, 39, 209–251. [Google Scholar] [CrossRef]
- Kubota, Y.; Koyama, Y.; Yamada, T.; Inagaki, S.; Tatsumi, T. Synthesis and catalytic performance of Ti-MCM-68 for effective oxidation reactions. Chem. Commun. 2008, 6224–6226. [Google Scholar] [CrossRef]
- Dorset, D.L.; Weston, S.C.; Dhingra, S.S. Crystal structure of zeolite MCM-68: A new three-dimensional framework with large pores. J. Phys. Chem. B 2006, 110, 2045–2050. [Google Scholar] [CrossRef]
- Koyama, Y.; Ikeda, T.; Tatsumi, T.; Kubota, Y. A multi-dimensional microporous silicate that is isomorphous to zeolite MCM-68. Angew. Chemie Int. Ed. 2008, 47, 1042–1046. [Google Scholar] [CrossRef]
- Sasaki, M.; Sato, Y.; Tsuboi, Y.; Inagaki, S.; Kubota, Y. Ti-YNU-2: A microporous titanosilicate with enhanced catalytic performance for phenol oxidation. ACS Catal. 2014, 4, 2653–2657. [Google Scholar] [CrossRef]
- Deka, R.C.; Nasluzov, V.A.; Ivanova Shor, E.A.; Shor, A.M.; Vayssilov, G.N.; Rösch, N. Comparison of all sites for Ti substitution in zeolite TS-1 by an accurate embedded-cluster method. J. Phys. Chem. B 2005, 109, 24304–24310. [Google Scholar] [CrossRef] [PubMed]
- Lamberti, C.; Bordiga, S.; Zecchina, A.; Artioli, G.; Marra, G.; Spanò, G. Ti location in the MFI framework of Ti-silicalite-1: A neutron powder diffraction study. J. Am. Chem. Soc. 2001, 123, 2204–2212. [Google Scholar] [CrossRef] [PubMed]
- Grand, J.; Talapaneni, S.N.; Vicente, A.; Fernandez, C.; DIb, E.; Aleksandrov, H.A.; Vayssilov, G.N.; Retoux, R.; Boullay, P.; Gilson, J.P.; et al. One-pot synthesis of silanol-free nanosized MFI zeolite. Nat. Mater. 2017, 16, 1010–1015. [Google Scholar] [CrossRef]
- Reddy, J.S.; Kumar, R.; Ratnasamy, P. Titanium silicalite-2: Synthesis, characterization and catalytic properties. Appl. Catal. 1990, 58, L1–L4. [Google Scholar] [CrossRef]
- Vayssilov, G.N.; Van Santen, R.A. Catalytic activity of titanium silicalites-A DFT study. J. Catal. 1998, 175, 170–174. [Google Scholar] [CrossRef]
- Nandi, P.; Tang, W.; Okrut, A.; Kong, X.; Hwang, S.J.; Neurock, M.; Katz, A. Catalytic consequences of open and closed grafted Al(III)-calix[4]arene complexes for hydride and oxo transfer reactions. Proc. Natl. Acad. Sci. USA 2013, 110, 2484–2489. [Google Scholar] [CrossRef] [Green Version]
- Wells, D.H.; Delgass, W.N.; Thomson, K.T. Evidence of Defect-Promoted Reactivity for Epoxidation of Propylene in Titanosilicate (TS-1) Catalysts: A DFT Study. J. Am. Chem. Soc. 2004, 126, 2956–2962. [Google Scholar] [CrossRef]
- Bonino, F.; Damin, A.; Ricchiardi, G.; Ricci, M.; Spanò, G.; D’Aloisio, R.; Zecchina, A.; Lamberti, C.; Prestipino, C.; Bordiga, S. Ti-peroxo species in the TS-1/H2O2/H2O system. J. Phys. Chem. B 2004, 108, 3573–3583. [Google Scholar] [CrossRef]
- Ikeda, T.; Inagaki, S.; Hanaoka, T.A.; Kubota, Y. Investigation of Si atom migration in the framework of MSE-type zeolite YNU-2. J. Phys. Chem. C 2010, 114, 19641–19648. [Google Scholar] [CrossRef]
- Jentys, A.; Catlow, C.R.A. Structural properties of titanium sites in Ti-ZSM5. Catal. Lett. 1993, 22, 251–257. [Google Scholar] [CrossRef]
- Millini, R.; Perego, G.; Seiti, K. Ti Substitution in MFI Type Zeolites: A Quantum Mechanical Study. Stud. Surf. Sci. Catal. 1994, 84, 2123–2129. [Google Scholar] [CrossRef]
- Petkov, P.S.; Aleksandrov, H.A.; Valtchev, V.; Vayssilov, G.N. Framework stability of heteroatom-substituted forms of extra-large-pore Ge-silicate molecular sieves: The case of ITQ-44. Chem. Mater. 2012, 24, 2509–2518. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metalamorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Jeanvoine, Y.; Ángyán, J.G.; Kresse, G.; Hafner, J. Brønsted acid sites in HSAPO-34 and chabazite: An Ab initio structural study. J. Phys. Chem. B 1998, 102, 5573–5580. [Google Scholar] [CrossRef]
- Hirata, S.; Head-Gordon, M. Time-dependent density functional theory within the Tamm-Dancoff approximation. Chem. Phys. Lett. 1999, 314, 291–299. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Dauenhauer, P.J.; Abdelrahman, O.A. A Universal Descriptor for the Entropy of Adsorbed Molecules in Confined Spaces. ACS Cent. Sci. 2018, 4, 1235–1243. [Google Scholar] [CrossRef]
- Ochterski, J.W. Thermochemistry in Gaussian; Gaussian Inc.: Wallingford, CT, USA, 2000; pp. 1–19. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T-Sites | ∆E V(Si) | ∆E(Ti → Si) | ∆E(Ti → Si vac) | ∆E(Al → Si) |
|---|---|---|---|---|
| T1 | −192 | −193 | −1 | −221 |
| T2 | −187 | −178 | 9 | −206 |
| T3 | −171 | −160 | 11 | −191 |
| T4 | −191 | −174 | 17 | −214 |
| T5 | −174 | −163 | 11 | −208 |
| T6 | −196 | −179 | 17 | −211 |
| T7 | −154 | −165 | −11 | −201 |
| T8 | −193 | −168 | 20 | −221 |
| T7T8 next | −211 | |||
| T7T8 far | −189 | |||
| T3T5 next | −257 | |||
| T3T5 far | −215 | |||
| MOR | −14 | |||
| ITQ−44 | −59 | |||
| MFI [13] | ~−90 |
| Zeo[(SiO2)111(TiO2)] + 4H2O → Zeo[(SiO2)110(TiO2)H4] + Si(OH)4 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Silanol Nest Position, ∆E [kJ/mol] | ||||||||
| Ti Position | T1 | T2 | T3 | T4 | T5 | T6 | T7 | T8 |
| Ti in T1 | −10 | 1 | −1 | −21 | ||||
| Ti in T2 | −29 | −38 | −20 | −26 | ||||
| Ti in T3 | −22 | −6 | −63 | −25 | ||||
| Ti in T4 | −48 | −43 | −9 | −46 | ||||
| Ti in T5 | −36 | −21 | −53 | −17 | ||||
| Ti in T6 | −25 | −16 | −13 | |||||
| Ti in T7 (MFI) 1 | 13 | −31 | −46 | −5 | ||||
| BE | ΔG | Ti-N | νcalc | νcalc | νcalc | νcalc | Δν | Δν | Δν | Δν | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Pyridine | 1567 | 1565 | 1454 | 1420 | |||||||
| T1_pyr | −101 | −65 | 239 | 1604 | 1562 | 1471 | 1425 | 37 | −3 | 17 | 5 |
| T2_pyr | −101 | −64 | 235 | 1590 | 1574 | 1468 | 1431 | 23 | 9 | 14 | 11 |
| T3_pyr | −95 | −55 | 239 | 1607 | 1568 | 1471 | 1424 | 40 | 3 | 17 | 4 |
| T4_pyr | −95 | −61 | 237 | 1589 | 1571 | 1469 | 1431 | 22 | 6 | 15 | 11 |
| T5_pyr | −103 | −63 | 235 | 1591 | 1568 | 1472 | 1433 | 24 | 3 | 18 | 13 |
| T6_pyr | −124 | −87 | 232 | 1611 | 1561 | 1466 | 1427 | 44 | −4 | 12 | 7 |
| T6_pyr_vac | −121 | −85 | 234 | 1606 | 1564 | 1464 | 1426 | 39 | −1 | 10 | 6 |
| CH3CN | 2297 | ||||||||||
| T1_CH3CN | −60 | −31 | 239 | 2335 | 38 | ||||||
| T2_CH3CN | −52 | −24 | 245 | 2325 | 28 | ||||||
| T3_CH3CN | −60 | −34 | 239 | 2327 | 30 | ||||||
| T4_CH3CN | −49 | −24 | 241 | 2331 | 34 | ||||||
| T5_CH3CN | −67 | −33 | 237 | 2338 | 41 | ||||||
| T6_CH3CN | −73 | −40 | 231 | 2341 | 44 | ||||||
| T6_CH3CN_vac | −45 | −16 | 234 | 2341 | 44 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petkov, P.S.; Simeonova, K.; Koleva, I.Z.; Aleksandrov, H.A.; Kubota, Y.; Inagaki, S.; Valtchev, V.; Vayssilov, G.N. Defect Formation, T-Atom Substitution and Adsorption of Guest Molecules in MSE-Type Zeolite Framework—DFT Modeling. Molecules 2021, 26, 7296. https://doi.org/10.3390/molecules26237296
Petkov PS, Simeonova K, Koleva IZ, Aleksandrov HA, Kubota Y, Inagaki S, Valtchev V, Vayssilov GN. Defect Formation, T-Atom Substitution and Adsorption of Guest Molecules in MSE-Type Zeolite Framework—DFT Modeling. Molecules. 2021; 26(23):7296. https://doi.org/10.3390/molecules26237296
Chicago/Turabian StylePetkov, Petko St., Kristina Simeonova, Iskra Z. Koleva, Hristiyan A. Aleksandrov, Yoshihiro Kubota, Satoshi Inagaki, Valentin Valtchev, and Georgi N. Vayssilov. 2021. "Defect Formation, T-Atom Substitution and Adsorption of Guest Molecules in MSE-Type Zeolite Framework—DFT Modeling" Molecules 26, no. 23: 7296. https://doi.org/10.3390/molecules26237296