
Abstract
Traumatic brain injury (TBI) is a leading cause of death and disability worldwide. The finding that cellular microparticles (MPs) generated by injured cells profoundly impact on pathological courses of TBI has paved the way for new diagnostic and therapeutic strategies. MPs are subcellular fragments or organelles that serve as carriers of lipids, adhesive receptors, cytokines, nucleic acids, and tissue-degrading enzymes that are unique to the parental cells. Their sub-micron sizes allow MPs to travel to areas that parental cells are unable to reach to exercise diverse biological functions. In this review, we summarize recent developments in identifying a casual role of MPs in the pathologies of TBI and suggest that MPs serve as a new class of therapeutic targets for the prevention and treatment of TBI and associated systemic complications.
Similar content being viewed by others


Introduction
Traumatic brain injury (TBI) is a leading cause of death and disability among adolescent males and young adults. Approximately 3.5 million TBI cases are reported each year in emergency rooms throughout the United States, resulting in approximately 50,000 annual deaths (Cuthbert et al., 2015; He et al., 2005). TBI undergoes two distinct, but partially overlapping phases of primary and secondary injures. The primary injury occurs at the moment of trauma as a result of mechanical forces that disrupt the structural integrity of the brain. It rapidly evolves into secondary biochemical and cellular changes (Ghajar, 2000). The interplay between the brain and other organs propagates oxidative, hemostatic, ischemic, and inflammatory injuries secondary to TBI. Cellular microparticles produced by injured tissues have been increasingly recognized as a key mediator for the interplay, promoting a transition from primary injury to secondary injury (Maas et al., 2010; Stoica and Faden, 2010). This review focuses on systemic impacts of cellular microparticles released from traumatized brains, with specific emphasis on TBI-associated coagulopathy (TBI-AC) and inflammation.
Cellular microparticles
MPs are a class of subcellular vesicles that are composed of shed membrane fragments and intracellular organelles and nuclear components. They are produced from cells undergoing active microvesiculation (Siljander et al., 2001; Heemskerk et al., 1997; Alkhamis et al., 1990; Owens and Mackman, 2011) or apoptosis (Shcherbina and Remold-O’Donnell, 1999; Dale and Friese, 2006; Brown et al., 2000). The former is triggered by the activation of the cysteine protease calpain, which disrupts the membrane-cytoskeleton association (Fox et al., 1991; Fox et al., 1990; Saatman et al., 2010; Zetterberg et al., 2013). This is a self-propelling mechanism whereby increasing intracellular Ca2+ induces glutamate release to activate the N-methyl-D-aspartate (NMDA) receptor, leading to further increases in cytosolic and mitochondrial Ca2+ levels (Cheng et al., 2012). The current definition of MPs is rather vague, describing them as heterogeneous particles of less than 1 μm in diameter from various types of cells (Owens and Mackman, 2011; Hugel et al., 2005; Cocucci et al., 2009). Their subcellular sizes allow MPs to travel to areas where the parental cells are unable to go (e.g., extracellular spaces such as those between endothelial cells). Furthermore, the differences in their surface molecules and carried cargoes allow MPs from different lineages of parental cells to manifest diverse biological activities.
MPs shed from cell membranes are enriched in microdomains (lipid rafts), where cholesterol, phospholipids, and functional receptors are clustered (Davizon et al., 2010; Biro et al., 2005). These surface molecules often serve as signatures of the parental cells (Moskovich and Fishelson, 2007). For example, platelet MPs express the adhesion receptor glycoprotein (GP) Ib-IX-V complex, the integrin αIIbβ3, and GPVI (Horstman et al., 2004). Endothelial MPs contain e-selectin (CD62e), cadherin 5 type 2 (CD144), and endoglin (CD105) (Horstman et al., 2004; Jimenez et al., 2003). MPs arising from the lipid rafts of monocytes contain tissue factor and P-selectin glycoprotein ligand 1 (Del Conde et al., 2005). MPs can also contain genomic and mitochondrial DNA and a variety of RNA species (ribosomal, messenger, and micro) that become MP-bound during cell apoptosis (Boudreau et al., 2014; Zhao et al., 2016; Miranda et al., 2010; Cai et al., 2013; Hasselmann et al., 2001; Reich and Pisetsky, 2009).
MPs can also be intracellular granules. For example, enzyme-rich lysosomes are detected in the circulation as CD68+ or CD63+ MPs (Horstman et al., 2004). We have recently detected intact or partially damaged mitochondria as either free microparticles or those embedded in plasma membrane of parental cells in the peripheral blood samples of mice subjected to severe TBI (Fig. 1) (Zhao et al., 2016). These mitochondrial microparticles (mtMPs) account for >55% of all annexin V-binding MPs found in the peripheral blood of mice subjected to acute TBI. The membrane integrity and internal structures of mtMPs are well maintained. This predominant presence of mtMPs is consistent with neurons and glial cells being mitochondria-rich to meet the high energy needs of brain cells.

mtMPs were detected in plasma samples from FPI mice. TEM images of mitochondria-free MPs (A), naked mitochondria (B), and mitochondria-embedded BDMP (C*: mitochondria) detected in plasma samples of FPI mice. (D) Mitochondria-lacking MPs, mitochondria (E), and mitochondria-embedded membrane microparticles (F*: mitochondria) from BDMPs produced in vitro. (G) The section of a non-injured mouse brain shows a dense perinuclear accumulation of mitochondria (left, arrowhead: nuclear membrane). A locally enlarged image further shows mitochondria (*), endoplasmic reticulum (ER) and membrane-bound, and free polyribosomes (arrow)
Because of their high energy consumption, brain cells are prone to the production of MPs as a result of ischemic and inflammatory insults secondary to TBI (Bianco et al., 2005; Ferrari et al., 1997; Duan and Neary, 2006). We have shown that more than 70,000/μL of brain-derived neuronal and glial cell microparticles (BDMPs) are detected in the peripheral blood of mice within hours after they are subjected to fluidic percussion injury (Tian et al., 2015). Our finding is consistent with other reports on elevated levels of MPs of various cell types in blood samples of TBI patients and animals subjected to TBI (Table 1). These BDMPs play a critical role in the development of TBI-associated coagulopathy defined as the inability of blood to clot.
Microparticles and TBI-associated coagulopathy
Coagulopathy often develops in trauma patients, primarily due to substantial blood losses (hemorrhagic shock), hemodilution after substantial transfusion of crystalloids, and hypothermia (Maani et al., 2009; Wafaisade et al., 2010). It is also reported in 10% to 97.2% of TBI patients, depending on the tests used to define the coagulopathy (Harhangi et al., 2008), even though isolated TBI lacks two critical triggers of coagulopathy—a large volume of blood loss and substantial fluid resuscitation (Chang et al., 2016; Zhang et al., 2012)—suggesting that TBI-AC follows a distinct pathway that differs from coagulopathy found in trauma to the trunk and limbs (Zhang et al., 2012; Corps et al., 2015). However, despite extensive studies in the past, the pathogenesis of TBI-AC remains poorly defined.
We have recently shown that BDMPs are a major mediator of TBI-AC (Tian et al., 2015). BDMPs released into the systemic circulation induce a rapid and systemic hypercoagulable state that is quickly turned into consumptive coagulopathy, consistent with clinical observations of patients with TBI-AC (Hulka et al.,1996; Stein and Smith, 2004). This phenotype observed in TBI mice is reproduced in uninjured mice that have been infused with purified BDMPs, implicating an essential role of MPs in the development of coagulopathy. The procoagulant activity of BDMPs is mediated by the procoagulant anionic phospholipids, primarily phosphatidylserine (PS), that are highly enriched on the surface of BDMPs. PS is normally present on the inner leaflet (Kunzelmann-Marche et al., 2000), whereas neutral phospholipids (e.g., phosphatidylcholine) are found on the external leaflet of a membrane bilayer (Phillis et al., 2006). This asymmetry is maintained by active transporters (Devaux, 1992; Suzuki et al., 2010), but is lost in cells undergoing apoptosis or microvesiculation, leading to the exposure of PS on the outer membrane (Zwaal et al., 1977). This PS-mediated procoagulant activity is expected to be higher for BDMPs because phospholipids are highly enriched on the membrane of brain cells (Sparvero et al., 2010), accounting for ~25% of the dry weight of an adult brain compared to ~10% of other tissues (Lentz, 2003; Nesheim and Mann, 1983). We further determined that cardiolipin (CL, (sn-3’-phosphatidyl)-sn-glycerol)), which is almost exclusively located in the mitochondrial inner membrane of a normal cell (Hovius et al., 1021; de Kroon et al., 1997), is exposed on the surface of mtMPs released from traumatized brain cells (Zhao et al., 2016). It is the exposed CL that makes mtMPs procoagulant at a level comparable to that of BDMPs. Our study also suggests that the procoagulant activity of anionic phospholipids requires their proper orientation and interaction with other membrane proteins as CL exposed on mtMPs was 1,600 times as active in promoting coagulation as an equivalent number of carrier-free CL micelles (Zhao et al., 2016). The procoagulant activity of microparticles is unlikely to be limited to neuron- and glial cell-derived MPs, as PS is ubiquitously distributed on the surface of all apoptotic cells (Nagata et al., 2010). For example, PS exposed on activated platelets also makes platelet-derived MPs procoagulant (Owens and Mackman, 2011; Ding et al., 2015). These platelet-derived MPs have been detected at elevated levels in patients with TBI (Nekludov et al., 2014; Morel et al., 2008; Jacoby et al., 2001), potentially contributing to the development of TBI-AC.
In addition to their direct influence on coagulation, BDMPs and mtMPs may also bind and activate platelets as they synergize with them to disrupt the integrity of the endothelial cell junctions, thereby allowing their release into the systemic circulation (Zhao et al., 2016; Tian et al., 2015). Platelet activation has indeed been reported during the acute phase of TBI (Jacoby et al., 2001; Awasthi et al., 1991; Auer and Ott, 1979). Because of the anionic phospholipid exposure, activated platelets and platelet-derived microparticles serve as platforms to promote systemic coagulation and thrombosis (Warren and Vales, 1972). The latter is characterized by platelet-rich and fibrin-rich intravascular microthrombosis that has been reported in TBI patients (Stein and Smith, 2004; Maeda et al., 1997; Kaufman et al., 1984; Steinet al., 2004) as well as in mouse models of TBI (Zhao et al., 2016; Tian et al., 2015; Maeda et al., 1997; van der Sande et al., 1981; Stein et al., 2002; Lu et al., 2004). Consistent with this notion, platelet dysfunctions have been reported to play a causal role in TBI-AC and TBI mortality (Schnuriger et al., 2010; Nekludov et al., 2007). It is very likely that these BDMPs interact with not only platelets (Tian et al., 2015), but also ECs and other cells, to spread and exaggerate coagulation, a key feature of consumptive coagulopathy.
The detection of substantial mtMPs in the circulation of mice subjected to TBI also raises an important question regarding the oxidative modification of phospholipids and proteins. If mtMPs released into the circulation remain active in ATP production, they could produce reactive oxygen species (ROS), the byproducts of ATP production (Murphy, 2009). These redox competent mtMPs may therefore serve as a source of oxidative stress and explain why CL is a preferred oxidation substrate in the condition of TBI (Ji et al., 2012). In fact, brain phospholipids are highly susceptible to oxidative stress (Bayir et al., 2007; Tyurin et al., 2008; Huvaere et al., 2010). The lipid peroxidation markers malondialdehyde and F2-isoprostane are increased in brain tissue, serum, and CSF after TBI (Sparvero et al., 2010; Seifman et al., 2008; Hoffman et al., 1996) and are associated with poor clinical outcomes (Pilitsis et al., 2003; Kasprzak et al., 2001). Whether oxidized phospholipids are more or less active in promoting coagulation remains a subject of further investigation, but the peroxidation of membrane phospholipids has been implicated in apoptosis, mitochondrial dysfunction, and various disease states (Bochkov et al., 2010; Maki et al., 2009; Frostegard et al., 2005). Oxidative stress is also a hallmark of TBI-induced inflammation that plays a major role in the development of secondary injury to the brain and other organs after TBI (Visavadiya et al., 2016; Hiebert et al., 2015; Cavallucci et al., 2014).
Microparticles and TBI-induced inflammation
Because of their prominent presence, redox-competent mtMPs could be a major source of oxidative stress that activates platelets (Arthur et al., 2008; Begonja et al., 2005), endothelial cells (Pearlstein et al., 2002; Li et al., 1999), neurons, glial cells (Visavadiya et al., 2016; Hiebert et al., 2015; Cavallucci et al., 2014), and the immune system (Dong, 2014) to promote inflammation. In support of this notion, multiple species of oxidized CL act as death signals for neurons as they induce the release of proapoptotic factors such as cytochrome C into the cytosol to activate caspases (Petrosillo et al., 2006; Kagan et al., 2005). This CL-induced membrane permeabilization involves several proteins on the outer membranes of mitochondria (Korytowski et al., 2011; Rostovtseva et al., 2006; Betaneli et al., 2012), consistent with our observation that CL on mtMPs is more procoagulant than purified CL micelles of equivalent concentrations. Free mitochondria released from platelets have been identified as a substrate for secreted phospholipase A2 group IIA (Boudreau et al., 2014), which is secreted by cells during the acute phase reaction (Birts et al., 2010). This phospholipase hydrolyzes the sn-2 acyl bond of glycerophospholipids to release free fatty acids and lysophospholipids to activate platelets and promote inflammation. Consistent with this proinflammatory activity, mitochondria that are intravenously injected into mice promote neutrophil activation and interaction with the endothelium (Boudreau et al., 2014).
MPs can significantly affect the immune system to propagate inflammation. MPs shed from neutrophils bind to bacteria through clusters of complement receptor 1 (Cr1), providing a robust defense against bacteria (Gasser et al., 2003; Hess et al., 1999). The Cr1 clusters are co-localized with myeloperoxidase and leukocyte elastase on the surface of MPs (Gasser et al., 2003; Hess et al., 1999), suggesting the involvement of oxidative actions during the activation of the immune system. During the acute phase reaction, neutrophil-derived MPs stimulate macrophages to release anti-inflammatory factors, such as transforming growth factor (TGF)-β, which, together with exposed PS on the surface of MPs, down-regulates inflammation (Gasser and Schifferli, 2004). By releasing anaphylatoxins upon the activation of the complement system, MPs also stimulate the host defense to initiate self-clearance through phagocytosis, thus eliminating MP-bound cytotoxin and proinflammatory mediators (Nauta et al., 2002). MPs can also carry and release the proinflammatory mediators such as IL-6, IL-1β, and CC-chemokine ligand (CCL)-2 (MCP-1) (MacKenzie et al., 2001; Mesri and Altieri, 1999), as well as vehicles to transfer chemokine receptors, such as CC-chemokine receptor 5 (CCR5) and CXC chemokine receptor 4 (CXCR4), between cells (Mack et al., 2000; Rozmyslowicz et al., 2003).
Microparticles from brain cells also have inflammation-promoting activities (Kumar and Loane, 2012; Obermeier et al., 2013). Microglia cells, which act as immune cells of the CNS, are triggered to microvesiculate when ATP binds to its receptor P2X7 on these cells (Bianco et al., 2005). These microglia-derived MPs contain the proprotein of the proinflammatory cytokine interleukin-1β (IL-1β), which is released upon cleavage by its processing enzyme caspase 1 (Bianco et al., 2005). A high level of microglia-derived MPs has been found in the peripheral blood and cerebrospinal fluid of patients with acute multiple sclerosis (Verderio et al., 2012; Saenz-Cuesta et al., 2014), a demyelinating autoimmune disease. Similarly, mice with autoimmune encephalomyelitis develop localized inflammation at the site where microglia-derived MPs were injected (Verderio et al., 2012). The production and biological activity of microglial MPs in the pathological course of TBI remain to be investigated. Astrocytes undergo a very similar process to generate IL-1β-containing microparticles in response to ATP (Bianco et al., 2009). The process of converting pro-IL-1β to its mature form requires the activation of acid sphingomyelinase in the outer leaflet of the plasma membrane (Bianco et al., 2009), suggesting a collaborative interaction between proteins and membrane phospholipids on the surface of MPs. Astrocytes also release mtMPs in a calcium-dependent manner that involves CD38 and cyclic ADP ribose signal (Hayakawa et al., 2016). Interestingly, these astrocyte-derived mtMPs are able to migrate into adjacent neurons to improve cell survival after ischemic stroke (Hayakawa et al., 2016). The production and activity of neuronal MPs are far less understood. It has been shown that a high level of mitochondrial Ca2+ increases the production of ROS and the activation of caspases, leading to neuronal apoptosis to produce MPs (Cheng et al., 2012). Neuronal MPs thus produced are reported to contain miRNA-21, which stimulates neuroinflammation and propagates damages to neurons after TBI (Harrison et al., 2016). Following membrane depolarization, cortical neurons from a mature mammalian brain release exosome-like vesicles (Lachenal et al., 2011; Faure et al., 2006) that selectively bind to adjacent neurons (Chivet et al., 2014). Exosomes isolated from the medium of primary cortical neuron cultures contain abundant microRNAs and small RNAs such as miR-124a. These exosomes can be internalized by astrocytes, leading to an increase in cytoplasm of astrocyte miR-124a and excitatory amino acid transporter 2 (EAAT2, also known as rodent analog GLT1). The latter is an important mediator of glutamate uptake in the brain (Morel et al., 2013).
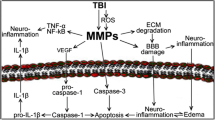
Finally, MPs and mtMPs synergize with platelets to disrupt the blood-brain barrier (BBB) (Zhao et al., 2016; Tian et al., 2015; Pan et al., 2016), which is composed of endothelial cells, pericytes, and astrocytes and is highly susceptible to traumatic and ischemic injures (Shetty et al., 2014). Since BBB regulates communications between systemic circulation and CNS, its disruption is therefore a key event that allows BDMPs to exert systemic influences. A broken endothelial barrier will also allow peripheral molecules to affect brain functions (Kumar and Loane, 2012; Shetty et al., 2014; Shlosberg et al., 2010). This brain-body interaction is sufficiently demonstrated in the development of BDMP-induced systemic coagulopathy (Fig. 2) (Zhao et al., 2016; Tian et al., 2015) .

A schematic illustration of MPs-mediated intercellular communication in TBI. Upon injury, neurons, microglia cells, and astrocytes release MPs into peripheral blood through disrupted BBB. These BDMPs activate platelets and promote platelet adhesion to the activated endothelium and exposed subendothelium at sites of vascular injury through multiple ligand-receptor interactions. Activated platelets also provide a PS-rich surface on which tissue factor forms a complex with coagulation factor VIIa to initiate the extrinsic pathway of coagulation. MPs interact with WBCs to promote inflammation
Conclusion and perspectives
MPs are an emerging class of biological mediators that share phenotypic and biological characteristics of their parental cells, while also engaging in activities that are distinct from those of their parental cells. MPs mediate intercellular communication (Zhao et al., 2016; Tian et al., 2015; Budnik et al., 2016; Zappulli et al., 2016), resulting in secondary injures such as systemic coagulopathy and inflammation after TBI (Zhao et al., 2016; Tian et al., 2015). In this regard, MPs serve as functional mediators for TBI-induced injuries and their progression. They could also serve as therapeutic targets of TBI and its secondary injuries. Because of the structural complexity and multi-level activities of MPs, there are outstanding questions and challenges in applying research to clinical diagnosis and therapeutics. The foremost challenge is to identify reliable and effective means of characterizing different types of MPs and distinguishing their functional specificities. The second is to dissect communications of MPs with cells that could lead to transient or persistent phenotypic changes of targeted cells.
References
Alkhamis TM, Beissinger RL, Chediak JR (1990) Artificial surface effect on red blood cells and platelets in laminar shear flow. Blood 75:1568–1575
Andrews AM, Lutton EM, Merkel SF, Razmpour R, Ramirez SH (2016) Mechanical injury induces brain endothelial-derived microvesicle release: implications for cerebral vascular injury during traumatic brain injury. Front Cell Neurosci 10:43
Arthur JF, Gardiner EE, Kenny D, Andrews RK, Berndt MC (2008) Platelet receptor redox regulation. Platelets 19:1–8
Auer LM, Ott E (1979) Disturbances of the coagulatory system in patients with severe cerebral trauma. II Platelet function. Acta Neurochir (Wien) 49:219–226
Awasthi D, Rock WA, Carey ME, Farrell JB (1991) Coagulation changes after an experimental missile wound to the brain in the cat. Surg Neurol 36:441–446
Bayir H, Tyurin VA, Tyurina YY, Viner R, Ritov V, Amoscato AA, Zhao Q, Zhang XJ, Janesko-Feldman KL, Alexander H, Basova LV, Clark RS, Kochanek PM, Kagan VE (2007) Selective early cardiolipin peroxidation after traumatic brain injury: an oxidative lipidomics analysis. Ann Neurol 62:154–169
Begonja AJ, Gambaryan S, Geiger J, Aktas B, Pozgajova M, Nieswandt B, Walter U (2005) Platelet NAD(P)H-oxidase-generated ROS production regulates alphaIIbbeta3-integrin activation independent of the NO/cGMP pathway. Blood 106:2757–2760
Betaneli V, Petrov EP, Schwille P (2012) The role of lipids in VDAC oligomerization. Biophys J 102:523–531
Bianco F, Pravettoni E, Colombo A, Schenk U, Moller T, Matteoli M, Verderio C (2005) Astrocyte-derived ATP induces vesicle shedding and IL-1 beta release from microglia. J Immunol 174:7268–7277
Bianco F, Perrotta C, Novellino L, Francolini M, Riganti L, Menna E, Saglietti L, Schuchman EH, Furlan R, Clementi E, Matteoli M, Verderio C (2009) Acid sphingomyelinase activity triggers microparticle release from glial cells. EMBO J 28:1043–1054
Biro E, Akkerman JW, Hoek FJ, Gorter G, Pronk LM, Sturk A, Nieuwland R (2005) The phospholipid composition and cholesterol content of platelet-derived microparticles: a comparison with platelet membrane fractions. J Thromb Haemost 3:2754–2763
Birts CN, Barton CH, Wilton DC (2010) Catalytic and non-catalytic functions of human IIA phospholipase A2. Trends Biochem Sci 35:28–35
Bochkov VN, Oskolkova OV, Birukov KG, Levonen AL, Binder CJ, Stockl J (2010) Generation and biological activities of oxidized phospholipids. Antioxid Redox Signal 12:1009–1059
Bohman LE, Riley J, Milovanova TN, Sanborn MR, Thom SR, Armstead WM (2016) Microparticles impair hypotensive cerebrovasodilation and cause hippocampal neuronal cell injury after traumatic brain injury. J Neurotrauma 33:168–174
Boudreau LH, Duchez AC, Cloutier N, Soulet D, Martin N, Bollinger J, Pare A, Rousseau M, Naika GS, Levesque T, Laflamme C, Marcoux G, Lambeau G, Farndale RW, Pouliot M, Hamzeh-Cognasse H, Cognasse F, Garraud O, Nigrovic PA, Guderley H, Lacroix S, Thibault L, Semple JW, Gelb MH, Boilard E (2014) Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood 124:2173–2183
Brown SB, Clarke MC, Magowan L, Sanderson H, Savill J (2000) Constitutive death of platelets leading to scavenger receptor-mediated phagocytosis. A caspase-independent cell clearance program. J Biol Chem 275:5987–5996
Budnik V, Ruiz-Canada C, Wendler F (2016) Extracellular vesicles round off communication in the nervous system. Nat Rev Neurosci 17:160–172
Cai J, Han Y, Ren H, Chen C, He D, Zhou L, Eisner GM, Asico LD, Jose PA, Zeng C (2013) Extracellular vesicle-mediated transfer of donor genomic DNA to recipient cells is a novel mechanism for genetic influence between cells. J Mol Cell Biol 5:227–238
Cavallucci V, Bisicchia E, Cencioni MT, Ferri A, Latini L, Nobili A, Biamonte F, Nazio F, Fanelli F, Moreno S, Molinari M, Viscomi MT, D’Amelio M (2014) Acute focal brain damage alters mitochondrial dynamics and autophagy in axotomized neurons. Cell Death Dis 5:e1545
Chang R, Cardenas JC, Wade CE, Holcomb JB (2016) Advances in the understanding of trauma-induced coagulopathy. Blood 128:1043–1049
Cheng G, Kong RH, Zhang LM, Zhang JN (2012) Mitochondria in traumatic brain injury and mitochondrial-targeted multipotential therapeutic strategies. Br J Pharmacol 167:699–719
Chivet M, Javalet C, Laulagnier K, Blot B, Hemming FJ, Sadoul R (2014) Exosomes secreted by cortical neurons upon glutamatergic synapse activation specifically interact with neurons. J Extracell Vesicles 3:24722
Cocucci E, Racchetti G, Meldolesi J (2009) Shedding microvesicles: artefacts no more. Trends Cell Biol 19:43–51
Corps KN, Roth TL, McGavern DB (2015) Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol 72:355–362
Cuthbert JP, Harrison-Felix C, Corrigan JD, Kreider S, Bell JM, Coronado VG, Whiteneck GG (2015) Epidemiology of adults receiving acute inpatient rehabilitation for a primary diagnosis of traumatic brain injury in the United States. J Head Trauma Rehabil 30:122–135
Dale GL, Friese P (2006) Bax activators potentiate coated-platelet formation. J Thromb Haemost 4:2664–2669
Davizon P, Munday AD, Lopez JA (2010) Tissue factor, lipid rafts, and microparticles. Semin Thromb Hemost 36:857–864
de Kroon AI, Dolis D, Mayer A, Lill R, de Kruijff B (1997) Phospholipid composition of highly purified mitochondrial outer membranes of rat liver and Neurospora crassa. Is cardiolipin present in the mitochondrial outer membrane? Biochem Biophys Acta 1325:108–116
Del Conde I, Shrimpton CN, Thiagarajan P, Lopez JA (2005) Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood 106:1604–1611
Devaux PF (1992) Protein involvement in transmembrane lipid asymmetry. Annu Rev Biophys Biomol Struct 21:417–439
Ding W, Kou J, Meng H, Kou Y, He Z, Cao M, Wang L, Bi Y, Thatte HS, Shi J (2015) Procoagulant activity induced by transcatheter closure of atrial septal defects is associated with exposure of phosphatidylserine on microparticles, platelets and red blood cells. Thromb Res 136:354–360
Dong JF (2014) Platelet microparticles are not created equal. Blood 124:2161–2162
Duan S, Neary JT (2006) P2X(7) receptors: properties and relevance to CNS function. Glia 54:738–746
Faure J, Lachenal G, Court M, Hirrlinger J, Chatellard-Causse C, Blot B, Grange J, Schoehn G, Goldberg Y, Boyer V, Kirchhoff F, Raposo G, Garin J, Sadoul R (2006) Exosomes are released by cultured cortical neurones. Mol Cell Neurosci 31:642–648
Ferrari D, Chiozzi P, Falzoni S, Dal Susino M, Collo G, Buell G, Di Virgilio F (1997) ATP-mediated cytotoxicity in microglial cells. Neuropharmacology 36:1295–1301
Fox JE, Austin CD, Boyles JK, Steffen PK (1990) Role of the membrane skeleton in preventing the shedding of procoagulant-rich microvesicles from the platelet plasma membrane. J Cell Biol 111:483–493
Fox JE, Austin CD, Reynolds CC, Steffen PK (1991) Evidence that agonist-induced activation of calpain causes the shedding of procoagulant-containing microvesicles from the membrane of aggregating platelets. J Biol Chem 266:13289–13295
Frostegard J, Svenungsson E, Wu R, Gunnarsson I, Lundberg IE, Klareskog L, Horkko S, Witztum JL (2005) Lipid peroxidation is enhanced in patients with systemic lupus erythematosus and is associated with arterial and renal disease manifestations. Arthritis Rheum 52:192–200
Gasser O, Schifferli JA (2004) Activated polymorphonuclear neutrophils disseminate anti-inflammatory microparticles by ectocytosis. Blood 104:2543–2548
Gasser O, Hess C, Miot S, Deon C, Sanchez JC, Schifferli JA (2003) Characterisation and properties of ectosomes released by human polymorphonuclear neutrophils. Exp Cell Res 285:243–257
Ghajar J (2000) Traumatic brain injury. Lancet 356:923–929
Harhangi BS, Kompanje EJ, Leebeek FW, Maas AI (2008) Coagulation disorders after traumatic brain injury. Acta Neurochir 150:165–175; discussion 175
Harrison EB, Hochfelder CG, Lamberty BG, Meays BM, Morsey BM, Kelso ML, Fox HS, Yelamanchili SV (2016) Traumatic brain injury increases levels of miR-21 in extracellular vesicles: implications for neuroinflammation. FEBS Open Bio 6:835–846
Hasselmann DO, Rappl G, Tilgen W, Reinhold U (2001) Extracellular tyrosinase mRNA within apoptotic bodies is protected from degradation in human serum. Clin Chem 47:1488–1489
Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, Ji X, Lo EH (2016) Transfer of mitochondria from astrocytes to neurons after stroke. Nature 535:551–555
He J, Gu D, Wu X, Reynolds K, Duan X, Yao C, Wang J, Chen CS, Chen J, Wildman RP, Klag MJ, Whelton PK (2005) Major causes of death among men and women in China. N Engl J Med 353:1124–1134
Heemskerk JW, Vuist WM, Feijge MA, Reutelingsperger CP, Lindhout T (1997) Collagen but not fibrinogen surfaces induce bleb formation, exposure of phosphatidylserine, and procoagulant activity of adherent platelets: evidence for regulation by protein tyrosine kinase-dependent Ca2+ responses. Blood 90:2615–2625
Hess C, Sadallah S, Hefti A, Landmann R, Schifferli JA (1999) Ectosomes released by human neutrophils are specialized functional units. J Immunol 163:4564–4573
Hiebert JB, Shen Q, Thimmesch AR, Pierce JD (2015) Traumatic brain injury and mitochondrial dysfunction. Am Journal Med Sci 350:132–138
Hoffman SW, Roof RL, Stein DG (1996) A reliable and sensitive enzyme immunoassay method for measuring 8-isoprostaglandin F2 alpha: a marker for lipid peroxidation after experimental brain injury. J Neurosci Methods 68:133–136
Horstman LL, Jy W, Jimenez JJ, Bidot C, Ahn YS (2004) New horizons in the analysis of circulating cell-derived microparticles. Keio J Med 53:210–230
Hovius R, Lambrechts H, Nicolay K, de Kruijff B (1021) Improved methods to isolate and subfractionate rat liver mitochondria. Lipid composition of the inner and outer membrane. Biochim Biophys Acta 1990:217–226
Hugel B, Martinez MC, Kunzelmann C, Freyssinet JM (2005) Membrane microparticles: two sides of the coin. Physiology (Bethesda) 20:22–27
Hulka F, Mullins RJ, Frank EH (1996) Blunt brain injury activates the coagulation process. Arch Surg 131:923–927; discussion 927–928
Huvaere K, Cardoso DR, Homem-de-Mello P, Westermann S, Skibsted LH (2010) Light-induced oxidation of unsaturated lipids as sensitized by flavins. J Phys Chem B 114:5583–5593
Jacoby RC, Owings JT, Holmes J, Battistella FD, Gosselin RC, Paglieroni TG (2001) Platelet activation and function after trauma. J Trauma 51:639–647
Ji J, Kline AE, Amoscato A, Samhan-Arias AK, Sparvero LJ, Tyurin VA, Tyurina YY, Fink B, Manole MD, Puccio AM, Okonkwo DO, Cheng JP, Alexander H, Clark RS, Kochanek PM, Wipf P, Kagan VE, Bayir H (2012) Lipidomics identifies cardiolipin oxidation as a mitochondrial target for redox therapy of brain injury. Nat Neurosci 15:1407–1413
Jimenez JJ, Jy W, Mauro LM, Horstman LL, Soderland C, Ahn YS (2003) Endothelial microparticles released in thrombotic thrombocytopenic purpura express von Willebrand factor and markers of endothelial activation. Br J Haematol 123:896–902
Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, Vlasova II, Zhao Q, Zou M, Di P, Svistunenko DA, Kurnikov IV, Borisenko GG (2005) Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol 1:223–232
Kasprzak HA, Wozniak A, Drewa G, Wozniak B (2001) Enhanced lipid peroxidation processes in patients after brain contusion. J Neurotrauma 18:793–797
Kaufman HH, Hui KS, Mattson JC, Borit A, Childs TL, Hoots WK, Bernstein DP, Makela ME, Wagner KA, Kahan BD et al (1984) Clinicopathological correlations of disseminated intravascular coagulation in patients with head injury. Neurosurgery 15:34–42
Korytowski W, Basova LV, Pilat A, Kernstock RM, Girotti AW (2011) Permeabilization of the mitochondrial outer membrane by Bax/truncated Bid (tBid) proteins as sensitized by cardiolipin hydroperoxide translocation: mechanistic implications for the intrinsic pathway of oxidative apoptosis. J Biol Chem 286:26334–26343
Kumar A, Loane DJ (2012) Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav Immun 26:1191–1201
Kunzelmann-Marche C, Satta N, Toti F, Zhang Y, Nawroth PP, Morrissey JH, Freyssinet JM (2000) The influence exerted by a restricted phospholipid microenvironment on the expression of tissue factor activity at the cell plasma membrane surface. Thromb Haemost 83:282–289
Lachenal G, Pernet-Gallay K, Chivet M, Hemming FJ, Belly A, Bodon G, Blot B, Haase G, Goldberg Y, Sadoul R (2011) Release of exosomes from differentiated neurons and its regulation by synaptic glutamatergic activity. Mol Cell Neurosci 46:409–418
Lentz BR (2003) Exposure of platelet membrane phosphatidylserine regulates blood coagulation. Prog Lipid Res 42:423–438
Li AE, Ito H, Rovira II, Kim KS, Takeda K, Yu ZY, Ferrans VJ, Finkel T (1999) A role for reactive oxygen species in endothelial cell anoikis. Circ Res 85:304–310
Lu D, Mahmood A, Goussev A, Qu C, Zhang ZG, Chopp M (2004) Delayed thrombosis after traumatic brain injury in rats. J Neurotrauma 21:1756–1766
Maani CV, DeSocio PA, Holcomb JB (2009) Coagulopathy in trauma patients: what are the main influence factors? Curr Opin Anaesthesiol 22:255–260
Maas AI, Roozenbeek B, Manley GT (2010) Clinical trials in traumatic brain injury: past experience and current developments. Neurotherapeutics 7:115–126
Mack M, Kleinschmidt A, Bruhl H, Klier C, Nelson PJ, Cihak J, Plachy J, Stangassinger M, Erfle V, Schlondorff D (2000) Transfer of the chemokine receptor CCR5 between cells by membrane-derived microparticles: a mechanism for cellular human immunodeficiency virus 1 infection. Nat Med 6:769–775
MacKenzie A, Wilson HL, Kiss-Toth E, Dower SK, North RA, Surprenant A (2001) Rapid secretion of interleukin-1beta by microvesicle shedding. Immunity 15:825–835
Maeda T, Katayama Y, Kawamata T, Aoyama N, Mori T (1997) Hemodynamic depression and microthrombosis in the peripheral areas of cortical contusion in the rat: role of platelet activating factor. Acta Neurochir Suppl 70:102–105
Maki RA, Tyurin VA, Lyon RC, Hamilton RL, DeKosky ST, Kagan VE, Reynolds WF (2009) Aberrant expression of myeloperoxidase in astrocytes promotes phospholipid oxidation and memory deficits in a mouse model of Alzheimer disease. J Biol Chem 284:3158–3169
Mesri M, Altieri DC (1999) Leukocyte microparticles stimulate endothelial cell cytokine release and tissue factor induction in a JNK1 signaling pathway. J Biol Chem 274:23111–23118
Midura EF, Jernigan PL, Kuethe JW, Friend LA, Veile R, Makley AT, Caldwell CC, Goodman MD (2015) Microparticles impact coagulation after traumatic brain injury. J Surg Res 197:25–31
Miranda KC, Bond DT, McKee M, Skog J, Paunescu TG, Da Silva N, Brown D, Russo LM (2010) Nucleic acids within urinary exosomes/microvesicles are potential biomarkers for renal disease. Kidney Int 78:191–199
Morel N, Morel O, Petit L, Hugel B, Cochard JF, Freyssinet JM, Sztark F, Dabadie P (2008) Generation of procoagulant microparticles in cerebrospinal fluid and peripheral blood after traumatic brain injury. J Trauma 64:698–704
Morel L, Regan M, Higashimori H, Ng SK, Esau C, Vidensky S, Rothstein J, Yang Y (2013) Neuronal exosomal miRNA-dependent translational regulation of astroglial glutamate transporter GLT1. J Biol Chem 288:7105–7116
Moskovich O, Fishelson Z (2007) Live cell imaging of outward and inward vesiculation induced by the complement c5b-9 complex. J Biol Chem 282:29977–29986
Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem J 417:1–13
Nagata S, Hanayama R, Kawane K (2010) Autoimmunity and the clearance of dead cells. Cell 140:619–630
Nauta AJ, Trouw LA, Daha MR, Tijsma O, Nieuwland R, Schwaeble WJ, Gingras AR, Mantovani A, Hack EC, Roos A (2002) Direct binding of C1q to apoptotic cells and cell blebs induces complement activation. Eur J Immunol 32:1726–1736
Nekludov M, Bellander BM, Blomback M, Wallen HN (2007) Platelet dysfunction in patients with severe traumatic brain injury. J Neurotrauma 24:1699–1706
Nekludov M, Mobarrez F, Gryth D, Bellander BM, Wallen H (2014) Formation of microparticles in the injured brain of patients with severe isolated traumatic brain injury. J Neurotrauma 31:1927–1933
Nesheim ME, Mann KG (1983) The kinetics and cofactor dependence of the two cleavages involved in prothrombin activation. J Biol Chem 258:5386–5391
Obermeier B, Daneman R, Ransohoff RM (2013) Development, maintenance and disruption of the blood-brain barrier. Nat Med 19:1584–1596
Owens AP III, Mackman N (2011) Microparticles in hemostasis and thrombosis. Circ Res 108:1284–1297
Pan Q, He C, Liu H, Liao X, Dai B, Chen Y, Yang Y, Zhao B, Bihl J, Ma X (2016) Microvascular endothelial cells-derived microvesicles imply in ischemic stroke by modulating astrocyte and blood brain barrier function and cerebral blood flow. Mol Brain 9:63
Patz S, Trattnig C, Grunbacher G, Ebner B, Gully C, Novak A, Rinner B, Leitinger G, Absenger M, Tomescu OA, Thallinger GG, Fasching U, Wissa S, Archelos-Garcia J, Schafer U (2013) More than cell dust: microparticles isolated from cerebrospinal fluid of brain injured patients are messengers carrying mRNAs, miRNAs, and proteins. J Neurotrauma 30:1232–1242
Pearlstein DP, Ali MH, Mungai PT, Hynes KL, Gewertz BL, Schumacker PT (2002) Role of mitochondrial oxidant generation in endothelial cell responses to hypoxia. Arterioscler Thromb Vasc Biol 22:566–573
Petrosillo G, Casanova G, Matera M, Ruggiero FM, Paradies G (2006) Interaction of peroxidized cardiolipin with rat-heart mitochondrial membranes: induction of permeability transition and cytochrome c release. FEBS Lett 580:6311–6316
Phillis JW, Horrocks LA, Farooqui AA (2006) Cyclooxygenases, lipoxygenases, and epoxygenases in CNS: their role and involvement in neurological disorders. Brain Res Rev 52:201–243
Pilitsis JG, Coplin WM, O’Regan MH, Wellwood JM, Diaz FG, Fairfax MR, Michael DB, Phillis JW (2003) Free fatty acids in cerebrospinal fluids from patients with traumatic brain injury. Neurosci Lett 349:136–138
Reich CF III, Pisetsky DS (2009) The content of DNA and RNA in microparticles released by Jurkat and HL-60 cells undergoing in vitro apoptosis. Exp Cell Res 315:760–768
Rostovtseva TK, Kazemi N, Weinrich M, Bezrukov SM (2006) Voltage gating of VDAC is regulated by nonlamellar lipids of mitochondrial membranes. J Biol Chem 281:37496–37506
Rozmyslowicz T, Majka M, Kijowski J, Murphy SL, Conover DO, Poncz M, Ratajczak J, Gaulton GN, Ratajczak MZ (2003) Platelet- and megakaryocyte-derived microparticles transfer CXCR4 receptor to CXCR4-null cells and make them susceptible to infection by X4-HIV. AIDS 17:33–42
Saatman KE, Creed J, Raghupathi R (2010) Calpain as a therapeutic target in traumatic brain injury. Neurotherapeutics 7:31–42
Saenz-Cuesta M, Irizar H, Castillo-Trivino T, Munoz-Culla M, Osorio-Querejeta I, Prada A, Sepulveda L, Lopez-Mato MP, Lopez de Munain A, Comabella M, Villar LM, Olascoaga J, Otaegui D (2014) Circulating microparticles reflect treatment effects and clinical status in multiple sclerosis. Biomark Med 8:653–661
Schnuriger B, Inaba K, Abdelsayed GA, Lustenberger T, Eberle BM, Barmparas G, Talving P, Demetriades D (2010) The impact of platelets on the progression of traumatic intracranial hemorrhage. J Trauma 68:881–885
Seifman MA, Adamides AA, Nguyen PN, Vallance SA, Cooper DJ, Kossmann T, Rosenfeld JV, Morganti-Kossmann MC (2008) Endogenous melatonin increases in cerebrospinal fluid of patients after severe traumatic brain injury and correlates with oxidative stress and metabolic disarray. J Cereb Blood Flow Metab 28:684–696
Shcherbina A, Remold-O’Donnell E (1999) Role of caspase in a subset of human platelet activation responses. Blood 93:4222–4231
Shetty AK, Mishra V, Kodali M, Hattiangady B (2014) Blood brain barrier dysfunction and delayed neurological deficits in mild traumatic brain injury induced by blast shock waves. Front Cell Neurosci 8:232
Shlosberg D, Benifla M, Kaufer D, Friedman A (2010) Blood-brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat Rev Neurol 6:393–403
Siljander P, Farndale RW, Feijge MA, Comfurius P, Kos S, Bevers EM, Heemskerk JW (2001) Platelet adhesion enhances the glycoprotein VI-dependent procoagulant response: involvement of p38 MAP kinase and calpain. Arterioscler Thromb Vasc Biol 21:618–627
Sparvero LJ, Amoscato AA, Kochanek PM, Pitt BR, Kagan VE, Bayir H (2010) Mass-spectrometry based oxidative lipidomics and lipid imaging: applications in traumatic brain injury. J Neurochem 115:1322–1336
Stein SC, Smith DH (2004) Coagulopathy in traumatic brain injury. Neurocrit Care 1:479–488
Stein SC, Chen XH, Sinson GP, Smith DH (2002) Intravascular coagulation: a major secondary insult in nonfatal traumatic brain injury. J Neurosurg 97:1373–1377
Stein SC, Graham DI, Chen XH, Smith DH (2004) Association between intravascular microthrombosis and cerebral ischemia in traumatic brain injury. Neurosurgery 54:687–691; discussion 691
Stoica BA, Faden AI (2010) Cell death mechanisms and modulation in traumatic brain injury. Neurotherapeutics 7:3–12
Suzuki J, Umeda M, Sims PJ, Nagata S (2010) Calcium-dependent phospholipid scrambling by TMEM16F. Nature 468:834–838
Tian Y, Salsbery B, Wang M, Yuan H, Yang J, Zhao Z, Wu X, Zhang Y, Konkle BA, Thiagarajan P, Li M, Zhang J, Dong JF (2015) Brain-derived microparticles induce systemic coagulation in a murine model of traumatic brain injury. Blood 125:2151–2159
Tschuor C, Asmis LM, Lenzlinger PM, Tanner M, Harter L, Keel M, Stocker R, Stover JF (2008) In vitro norepinephrine significantly activates isolated platelets from healthy volunteers and critically ill patients following severe traumatic brain injury. Crit Care 12:R80
Tyurin VA, Tyurina YY, Kochanek PM, Hamilton R, DeKosky ST, Greenberger JS, Bayir H, Kagan VE (2008) Oxidative lipidomics of programmed cell death. Methods Enzymol 442:375–393
van der Sande JJ, Emeis JJ, Lindeman J (1981) Intravascular coagulation: a common phenomenon in minor experimental head injury. J Neurosurg 54:21–25
Verderio C, Muzio L, Turola E, Bergami A, Novellino L, Ruffini F, Riganti L, Corradini I, Francolini M, Garzetti L, Maiorino C, Servida F, Vercelli A, Rocca M, Dalla Libera D, Martinelli V, Comi G, Martino G, Matteoli M, Furlan R (2012) Myeloid microvesicles are a marker and therapeutic target for neuroinflammation. Ann Neurol 72:610–624
Visavadiya NP, Patel SP, VanRooyen JL, Sullivan PG, Rabchevsky AG (2016) Cellular and subcellular oxidative stress parameters following severe spinal cord injury. Redox Biol 8:59–67
Wafaisade A, Wutzler S, Lefering R, Tjardes T, Banerjee M, Paffrath T, Bouillon B, Maegele M, Trauma Registry of D.G.U. (2010) Drivers of acute coagulopathy after severe trauma: a multivariate analysis of 1987 patients. Emerg Medicine Journal (EMJ) 27:934–939
Warren BA, Vales O (1972) The release of vesicles from platelets following adhesion to vessel walls in vitro. Br J Exp Pathol 53:206–215
Yasui H, Donahue DL, Walsh M, Castellino FJ, Ploplis VA (2016) Early coagulation events induce acute lung injury in a rat model of blunt traumatic brain injury. Am J Physiol Lung Cell Mol Physiol 311:L74–L86
Zappulli V, Friis KP, Fitzpatrick Z, Maguire CA, Breakefield XO (2016) Extracellular vesicles and intercellular communication within the nervous system. J Clin Invest 126:1198–1207
Zetterberg H, Smith DH, Blennow K (2013) Biomarkers of mild traumatic brain injury in cerebrospinal fluid and blood. Nat Rev Neurol 9:201–210
Zhang J, Jiang R, Liu L, Watkins T, Zhang F, Dong JF (2012) Traumatic brain injury-associated coagulopathy. J Neurotrauma 29:2597–2605
Zhao Z, Wang M, Tian Y, Hilton T, Salsbery B, Zhou EZ, Wu X, Thiagarajan P, Boilard E, Li M, Zhang J, Dong JF (2016) Cardiolipin-mediated procoagulant activity of mitochondria contributes to traumatic brain injury-associated coagulopathy in mice. Blood 127:2763–2772
Zwaal RF, Comfurius P, van Deenen LL (1977) Membrane asymmetry and blood coagulation. Nature 268:358–360
ACKNOWLEDGEMENTS
This study is supported by NIH grants NS087296 and HL71895 (JFD), National Natural Science Foundation of China State Key Program Grant 81330029 and National Natural Science Foundation of China research Grants 81271361, 81271359 (JNZ) and 81601068 (ZLZ).
ABBREVIATIONS
BBB, blood-brain barrier; BDMP, brain-derived microparticles; CCL, CC-chemokine ligand; CCR5, CC-chemokine receptor 5; CL, cardiolipin; Cr1, complement receptor 1; CSF, cerebrospinal fluid; CXCR4, CXC chemokine receptor 4; EAAT2, excitatory amino acid transporter 2; EMP, endothelial-derived microparticles; EV, extracellular vesicles; GFAP, glial fibrillary acidic protein; GP, glycoprotein; IL-1β, interleukin-1β; LMP, leukocyte-derived microparticles; miR, micro RNA; MP, microparticles; mtMPs, mitochondrial miroparticles; NMDA, N-methyl-D-aspartate; PMP, platelet-derived microparticles; PS, phosphatidylserine; ROS, reactive oxygen species; TBI, traumatic brain injury; TBI-AC, TBI-associated coagulopathy; TF, tissue factor; TGF-β, transforming growth factor-β
COMPLIANCE WITH ETHICS GUIDELINES
Zilong Zhao, Yuan Zhou, Ye Tian, Min Li, Jing-fei Dong, and Jianning Zhang declare that they have no conflict of interest. All institutional and national guidelines for the care and use of laboratory animals were followed.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zhao, Z., Zhou, Y., Tian, Y. et al. Cellular microparticles and pathophysiology of traumatic brain injury. Protein Cell 8, 801–810 (2017). https://doi.org/10.1007/s13238-017-0414-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13238-017-0414-6